Alzheimer's Disease (AD), the most common form of dementia, is a chronic neurodegenerative disease. The HECT domain and ankyrin repeat containing E3 ubiquitin protein ligase 1 (HACE1) gene is expressed in human brain and may play a role in the pathogenesis of neurodegenerative disorders. Till now, no previous study has reported the association of the HACE1 gene with the risk and Age at Onset (AAO) of AD; while few studies have checked the proportional hazards assumption in the survival analysis of AAO of AD using Cox proportional hazards model. In this study, we examined the associations of 14 Single Nucleotide Polymorphisms (SNPs) in the HACE1 gene with the risk and the AAO of AD using 791 AD patients and 782 controls. Multiple logistic regression model identified one SNP (rs9499937 with p = 1.8 × 10-3) to be associated with the risk of AD. For survival analysis of AAO, both classic Cox regression model and Bayesian survival analysis using the Cox proportional hazards model were applied to examine the association of each SNP with the AAO. The Hazards Ratio (HR) with its 95% Confidence Interval (CI) was estimated. Survival analysis using the classic Cox regression model showed that 4 SNPs were significantly associated with the AAO (top SNP rs9499937 with HR = 1.33, 95% CI = 1.13-1.57, p = 5.0 × 10-4). Bayesian Cox regression model showed similar but a slightly stronger associations (top SNP rs9499937 with HR = 1.34, 95% CI = 1.11-1.55) compared with the classic Cox regression model. Using an independent family-based sample, one SNP rs9486018 was associated with the risk of AD (p = 0.0323) and the T-T-G haplotype from rs9786015, rs9486018 and rs4079063 showed associations with both the risk and AAO of AD (p = 2.27 × 10-3 and 0.0487, respectively). The findings of this study provide first evidence that several genetic variants in the HACE1 gene were associated with the risk and AAO of AD.
Alzheimer's disease, Age at onset, HACE1, Single nucleotide polymorphisms, Survival analysis, Cox model, Bayesian analysis
Alzheimer's Disease (AD) is a chronic neurodegenerative disease [1]. It is estimated that, in 2010, about 4.7 million people in the United States (US) aged 65 years or older live with AD, and this number is projected to rise to 13.8 million, a nearly three-fold increase, by 2050 [2]. Weuve et al. [3] estimated the number of adults (aged ≥ 65 years) with AD in each US state and the District of Columbia (DC) and found that the number of older adults (aged ≥ 65 years) with AD in the US ranged from 5.1 (Alaska) to 530 million (California) in 2010; while in 2010, older adults with AD dementia comprised a median of 1.6% of a given state's total population; whereas by 2025, throughout the US, this proportion will keep increasing by nearly one-third on average [3]. The prevalence was estimated to be about 3.12% in 2012 for older adults (≥ 60 years) in the US using the Nationwide Inpatient Sample (NIS) 2002-2012 [4]. A meta-analysis estimated the prevalence for the combined population of African-Americans and Caucasians aged 65-90 in 2013 was 5.7% in the US; the prevalence for African-Americans aged 65-90 years was 8.6%, compared to 5.5% for Caucasians [5]. Globally, 26.6 million people (0.40% of the world population) suffered from AD in 2006 and it was predicted to affect 1 in 85 people by 2050 [6]. A recent meta-analysis showed that the prevalence of AD in Europe was 5.05% for adult (aged ≥ 50 years) (3.31% for males and 7.13% females) [7]. Increasing evidence suggests that autophagy may play a central role in AD [8,9]. The genetic heritability of AD ranges from 49% to 79% based on reviews of twin and family studies [10]. In addition to the risk, the Age at Onset (AAO) of AD has a genetic component with heritability about 42% [11,12].
The HECT domain and ankyrin repeat containing E3 ubiquitin protein ligase 1 (HACE1) gene (also known as KIAA1320) is located at 6q16.3 [13-15]. The HACE1 is expressed in brain, heart, lung, kidney, testis, and ovary [13,15]. Several studies have implicated that HACE1 is a candidate chromosome 6q21 tumor suppressor gene involved in multiple cancers [16-18]. Recently, it has been reported that HACE1 gene may play a role in neurodegeneration [19] and autophagy pathway [20]; while HACE1 mutations are involved in an autosomal recessive neurodevelopmental disorder [21], and glutamine addiction [22]. A Genome-Wide Association Study (GWAS) identified five Single-Nucleotide Polymorphisms (SNPs) (rs17065302, rs11759010, rs6927608, rs4946645 and rs4245525) within the HACE1 gene associated with equol-producing phenotype such as blood pressure, which may implicate HACE1 in intestinal immune responses [23]. Another GWAS identified 5 SNPs (rs4336470. rs9404576, rs4079063, rs24996663, and rs2499667) in HACE1 associated with neuroblastoma susceptibility [24]; while in a replication study, rs4336470 showed moderate association (p < 0.05) with risk of neuroblastoma [25]. More recently, it has been reported that the five above SNPs in the HACE1 gene may have a weak combined effect (p = 0.065) on neuroblastoma risk in Southern Chinese children [26]. Thus we hypothesized that HACE1 genetic variants may be in association with AD development.
To our best knowledge, no study has focused on the association of the HACE1 gene with the risk and AAO of AD. Even though the Cox proportional hazards model has been used to detect genetic associations with the AAO of AD [27-29], the proportional hazards assumption may be violated and they might not be carefully checked. Bayesian methods have been widely used recently in genetic association studies and provide alternative ways to traditional statistical methods [30-32]. In this study, we explored the association of HACE1 with the AAO of AD by using a Bayesian proportional hazards model in a population-based sample and then a family-based sample for replication.
791 patients with AD and 782 controls with complete genotype and phenotype information in a Canadian sample were selected from the Multi-Site Collaborative Study for Genotype-Phenotype Associations in Alzheimer's disease and the longitudinal follow-up of Genotype-Phenotype Associations in Alzheimer's disease and the Neuroimaging component of Genotype-Phenotype Associations in Alzheimer's disease-Study Accession: phs000219.v1.p1. Covariates include sex and age. The details about these subjects were described in previous studies [27,33]. Genotyping was conducted using the Affymetrix technique. The genotypes of 14 SNPs within the HACE1 gene were available in this data.
A family-based study (1266 AD cases and their relatives, 1070 individuals with the AAO values) were available from the National Institute on Aging - Late Onset Alzheimer's Disease (NIA-LOAD) Family Study: Genome-Wide Association Study for Susceptibility Loci - Study Accession: phs000168.v1.p1. Genotyping by the Center for Inherited Disease Research (CIDR) was performed using the Illumina Infinium II assay protocol. The details about the sample of subjects were described elsewhere [34]. There are 28 SNPs within the HACE1 gene in this family-based sample.
Descriptive statistics were used to characterize participants' sex, age and the AAO of AD stratified by AD case and control status [29,35]. Hardy-Weinberg Equilibrium (HWE) was tested for all SNPs using the controls by HAPLOVIEW software [36]. Then, Minor Allele Frequency (MAF) was determined for each SNP. Pair wise Linkage Disequilibrium (LD) statistics (r2) among SNPs were assessed using the European sample from the HapMap dataset (http://hapmap.ncbi.nlm.nih.gov/) and the founders in the family study.
Multiple logistic regression analysis of each SNP with the risk of AD as a binary outcome, adjusted for sex and age, was performed using PLINK [37]; while the asymptotic p-values were obtained and the Odds Ratio (OR) and 95% Confident Interval (CI) were estimated. The parallel procedure was performed for the multiple linear regression analysis of each SNP with the AAO of AD as a continuous outcome. Bonferroni correction (α = 0.05/14 = 3.57 × 10-3) was used to deal with the multiple comparison issue [38].
The Cox proportional hazards model (1) or Cox regression model [39] is widely used in the analysis of time-to-event data [40-42].
where is the hazard at time t for a subject (AAO for this study), is the baseline hazard function. The Hazard Ratio (HR) is defined as the ratio of the predicated hazard function under two different values of a predictor variable. The PHREG procedure in SAS fits the Cox model by maximizing the partial likelihood function. Both the graphical and numerical methods [43] were used to check the proportional hazards assumption in the ASSESS option of PROC PHREG. The ASSESS option plots the cumulative score residuals against time for each independent variable; while the RESAMPLE option computes the p-value of a Kolmogorov-type supremum test based on a sample of 1,000 simulated residual patterns. A significant p-value indicates a poor fit.
The Akaike Information Criterion (AIC) (2) was used as a measure of better fit among candidate models [44,45].
where x is the random variable, is the maximum likelihood estimate, and k is the number of parameters. A smaller AIC generally indicates a better fit.
Bayesian statistics is an extension of Bayes theorem, which can be written as (3)
where θ is the parameter of interest, Y is the observed evidence, P(Y) is the marginal probability, P(Y|θ) is the likelihood function, P(θ) is the prior, and P(θ|Y) is the posterior probability [32]. Bayesian Cox regression can be requested by using the BAYES statement in the PHREG procedure. Summary statistics (Mean, Standard Deviation, the Highest Posterior Density (HPD) and Credible Intervals, and Correlation Matrix) were computed for each of the parameters. Trace plots, posterior density plots, and autocorrelation function plots were also provided [32]. For Bayesian survival analysis of the AAO of AD, the normal prior was chosen for the coefficients and the Deviance Information Criteria (DIC) was available instead of AIC. DIC is intended as a generalization of AIC [46]. A measure of effective numbers of parameters is defined as pD in (4), where pD is the posterior mean deviance minus the deviance measured at the posterior mean of the parameters.
Then DIC is defined analagously to AIC as in (5). Models with smaller DIC are better supported by the data.
The PHREG procedure in SAS was used to fit the Cox model. Multiple Cox regression model analysis, adjusted for sex and age, was conducted to examine association of each SNP with the AAO of AD. Descriptive statistics and Cox regression analysis were performed with SAS v.9.4 (SAS Institute, Cary, NC, USA).
A family-based association analysis for AD was performed using PBAT version 3.6.7 [47]. For the risk of AD, the Family‐Based Association Test using Generalized Estimating Equations (FBAT-GEE) was used [48]; while for the AAO, FBAT-Wilcoxon statistics were employed [49]. The AAO values for healthy siblings were censored and age at entry into the study was used. Haplotype analysis was conducted in 2 or 3-SNP sliding window.
The demographic characteristics of the subjects are detailed in Table 1. The mean AAO for cases was 76.4 and 72.3 years, respectively, in the NIA and Canadian samples, respectively. All 14 SNPs had MAF > 5% and were in HWE in the controls (p > 0.05); while 1 of 28 SNPs with HWE < 0.10-4 in the family sample was removed for further analysis.
Table 1: Descriptive characteristics of cases and controls. View Table 1
We found that one SNP was associated with the risk of AD (rs9499937 with p = 1.8 × 10-3) and four SNPs were associated with the AAO of AD (rs7746856, rs6941988, rs9499937 and rs7770002 with p = 3.09 × 10-2, 3.88 × 10-3, 7.39 × 10-4, and 3.14 × 10-2, respectively) (Table 2). Interestingly, the same SNP rs9499937 showed associations with both the risk and AAO of AD and the results remained significant after Bonferroni correction (p < 3.57 × 10-3).
Table 2: SNPs associated with the risk and/or age at onset of AD (p < 0.05). View Table 2
Figure 1 and Figure 2 display the observed standardized score process with 20 simulated realizations from the null distribution for rs9499937 CC and CT genotypes, respectively. The plots showed that the observed process was atypical compared to the simulated realizations and revealed proportional hazards for the two genotypes compared with TT. The Kolmogorov-type supremum test results based on 1,000 simulations for all the covariates were not significant (p > 0.05), suggesting the proportional hazards assumption was valid for all the variables in the Canadian sample.
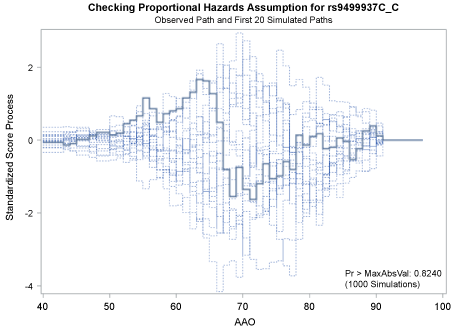 Figure 1: Explore plot for checking proportional hazards assumption for rs9499937C_C gentoype compared with rs9499937T_T genotype. View Figure 1
Figure 1: Explore plot for checking proportional hazards assumption for rs9499937C_C gentoype compared with rs9499937T_T genotype. View Figure 1
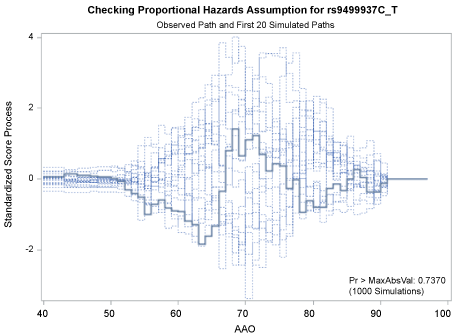 Figure 2: Explore plot for checking proportional hazards assumption for rs9499937C_T compared with rs9499937T_T.
View Figure 2
Figure 2: Explore plot for checking proportional hazards assumption for rs9499937C_T compared with rs9499937T_T.
View Figure 2
The classic Cox model showed that four SNPs were associated with the AAO of AD (the CT genotype of the top SNP rs9499937 with HR = 1.33, 95% CI = 1.13-1.57, p = 5.0 × 10-4). The HRs based on the Bayesian survival analyses revealed similar but a slightly stronger associations compared with the non-Bayesian analyses results (Table 3). The DIC for the four SNPs were similar to those of AIC. The trace plot, posterior density plot, and autocorrelation function plot based on Bayesian analysis (Figure 3) indicated that the Markov chain had stabilized with good mixing for rs9499937. The posterior density plot, which estimates the posterior marginal distributions for the four regression coefficients, showed a smooth and unimodal shape for the posterior marginal distribution (Figure 4).
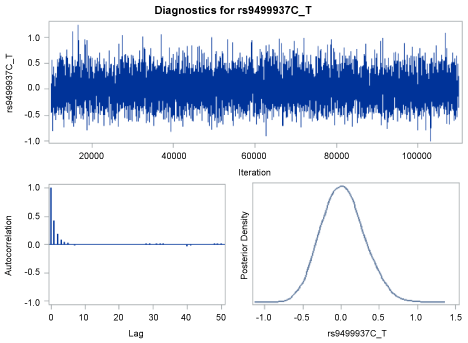 Figure 3: Trace plot, autocorrelation function plot, and posterior density plot for rs9499937. View Figure 3
Figure 3: Trace plot, autocorrelation function plot, and posterior density plot for rs9499937. View Figure 3
 Figure 4: The posterior density plots for the 4 regression coefficients. View Figure 4
Figure 4: The posterior density plots for the 4 regression coefficients. View Figure 4
Table 3: SNPs associated with the age at onset of AD using PROC PHREG (p < 0.05). View Table 3
We observed one SNP associated with the risk of AD (rs9486018 with p = 0.0323) by using FBAT-GEE analysis in the family-based study. The T-A haplotype from rs6937432 and rs6940552 revealed mostly significant associations with the risk (p = 9.7 × 10-4). The T-T haplotype from rs9786015 and rs9486018 and the T-G haplotype from rs9486018 and rs4079063 showed significant association with the risk (p = 1.56 × 10-3 and 4.46 × 10-3, respectively) (Table 4). Using the FBAT-Wilcoxon test, the C-C haplotype from rs6937026 and rs6946640 revealed mostly significant associations with the AAO (p = 0.0223). The C-A-G and C-C-A haplotype from rs6437026, rs4946640 and rs6910034 showed associations with the AAO (p = 0.0274 and 0.0225, respectively). The T-T-G haplotype from rs9786015, rs9486018 and rs4079063 showed associations with both the risk (p = 2.27 × 10-3) and the AAO (p = 0.0487).
Table 4: Haplotype analysis of the risk and age at onset of AD in the family sample. View Table 4
Using the HapMap data, we identified one haplotype block including the four SNPs associated AD and/or AAO in the case-control study. Figure 5 shows the LD structure based on LD statistics (r2). Based on the rough rule of thumb, values of r2 > 1/3 might indicate sufficiently strong LD that can be used for a fine mapping [50]. The neuroblastoma associated rs4336470 [24,25] had moderate or strong LD with three AAO associated SNPs (rs7746856, rs6941988 and rs7770002 with r2 = 0.6, 0.6 and 0.71, respectively) and weak LD with the risk and the AAO - associated SNP rs9499937 (r2 = 0.27). Furthermore, there was strong LD observed between rs2499663 and three AAO associated SNPs (rs7746856, rs6941988 and rs7770002 with r2 = 0.81, 0.81 and 0.93, respectively). Using the founders in the family study, LD structure based on (r2) was constructed for all 28 SNPs (Figure 6). The two neuroblastoma associated SNPs (rs4336470 and rs9404576) [24,25] had moderate LD with SNPs (such as rs6910034, rs9404573, rs9499934, rs696937432, rs9486015, and rs4079063) which built AD and AAO associated haplotype; while another neuroblastoma associated SNPs rs2499663 [24,25] had moderate or strong LD with SNPs involved in AD and AAO associated haplotype.
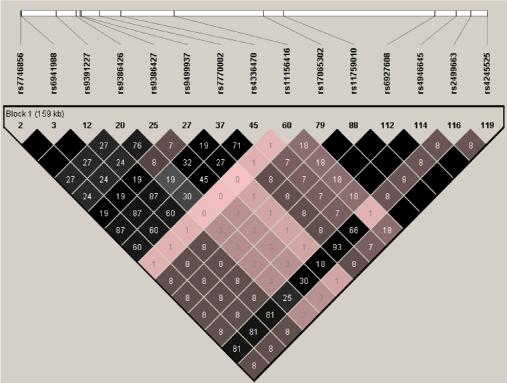 Figure 5: Linkage disequilibrium structure (r2) within the HACE1 gene using the HapMap data (Dark area shows r2 = 1). View Figure 5
Figure 5: Linkage disequilibrium structure (r2) within the HACE1 gene using the HapMap data (Dark area shows r2 = 1). View Figure 5
In the present study we explored the association of 14 HACE1 SNPs with the risk and AAO of AD using a case-control study and identified one SNP associated with AD and four SNPs with the AAO of AD using PLINK software. Interestingly, the same SNP rs9499937 showed associations with both the risk and AAO of AD. Bayesian Cox regressions revealed similar but a slightly stronger associations with the AAO of AD. Using an independent family-based sample, one SNP rs9486018 was associated with the risk of AD while haplotype analyses further revealed the associations with the risk and AAO of AD. The findings of this study provide the first evidence that several genetic variants in the HACE1 gene influenced the risk and the AAO of AD.
A previous study suggested that rs9391227 in the HACE1 gene was associated with celiac disease involved in the immune system and antigen presentation [51]; while another study revealed that five SNPs (rs17065302, rs11759010, rs6927608, rs4946645 and rs4245525) within HACE1 gene associated with equol-producing phenotype such as blood pressure [23] which may implicate HACE1 in immune responses. However, rs9391227 [51] was not available in both the case-control and family-based samples; while equol-producing phenotype associated five SNPs [23] were not associated with the risk or the AAO of AD in the case-control study and rs6927608 was not associated with the risk and AAO of AD in both samples. However, rs9391227 had strong LD with three AAO associated SNPs (rs7746856, rs6941988 and rs7770002 with r2 = 1.0, 1.0 and 0.87, respectively) in the case-control study (Figure 5); whereas rs6927608 had weak LD with other SNPs (Figure 5 and Figure 6). Previous studies have shown that AD is a chronic neurodegenerative disease while autophagy, immune and inflammatory processes are involved in the pathogenesis of AD [1,8,9,52-56].
 Figure 6: Linkage disequilibrium structure (r2) within the HACE1 gene of 28 SNPs in the family sample (Dark area shows r2 = 1). View Figure 6
Figure 6: Linkage disequilibrium structure (r2) within the HACE1 gene of 28 SNPs in the family sample (Dark area shows r2 = 1). View Figure 6
Previous epidemiology studies have suggested comorbidity of AD with certain cancers [57-59]; whereas there may be an inverse link between cancer and AD [59-61]. Recently, a meta-analysis of nine studies supports an association between AD and decreased cancer risk [62]. However, the mechanism still remains unclear [60]. Several SNPs (such as rs4336470, rs9404576, rs4079063, rs24996663, and rs2499667) in the HACE1 have been found to be associated with neuroblastoma susceptibility [24-26]. In the present study, the T-G haplotype inferred from the rs9486018 and rs4079063 was found to be associated with the risk of AD; while the T-T-A haplotype inferred from the rs9486015, rs9486018 and rs4079063 was found to be associated with the AAO of AD in the family-based study (Table 4). Furthermore, four neuroblastoma associated SNPs (rs4336470, rs9404576, rs4079063 and rs2499663) [24-26] had moderate to strong LDs with SNPs which built AD and AAO associated haplotypes (Figure 6). In addition, recently, it has been reported that HACE1 may play a role in neurodevelopment and addiction [19,21,22]. Taken together, the above findings may suggest that HACE1 gene may be involved in the pathogenesis of AD, cancers and blood pressure; however, the mechanism warrants further studies.
Several strengths of this study are worthy of noting. The present study provides the first evidence of several genetic variants within the HACE1 gene associated with the risk and the AAO of AD using a case-control sample and a family-based sample for replication. Furthermore, we checked the proportional hazards assumption using both the graphical and numerical methods for the Cox proportional hazards model and found that the proportional hazards assumption was valid for the AAO of AD data in the Canadian sample. In addition, we conducted Bayesian survival analysis of genetic variants with the AAO of AD. Bayesian method may provide an alternative approach to assessing and verifying associations that alleviates the limitations of p-values at the cost of some additional modeling and it has recently made great inroads in genetic association studies [30]. Like other research studies, some limitations also exist in this study. First, due to different genotyping platforms, only two SNPs (rs6927608 and rs24999663) overlap in two samples; therefore, our replication results in the family-based sample are gene-based rather than SNP-based. Second, only one SNP rs9499937 was associated with the risk and AAO of AD in the case-control study; while one haplotype T-T-G was associated with the risk and AAO of AD in the family-based study; which revealed heterogeneity between risk and AAO of AD. In addition, our current findings might be subject to type I error and need to be replicated in future studies.
To the best of our knowledge, this is the first candidate gene study which investigated the associations of HACE1 SNPs with the risk of and the AAO of AD. The findings may serve as a resource for replication in other populations for future investigations on target genetic variation and AD. Future functional studies of this gene may help better characterize the genetic architecture of the risk of and AAO of AD.
We acknowledge the NIH GWAS Data Repository, the Contributing Investigator(s) who contributed the phenotype data and DNA samples from his/her original study and the primary funding organization that supported the contributing study "Multi-Site Collaborative Study for Genotype-Phenotype Associations in Alzheimer's disease and longitudinal follow-up of Genotype-Phenotype Associations in Alzheimer's disease and Neuroimaging component of Genotype-Phenotype Associations in Alzheimer's disease" and "National Institute on Aging - Late Onset Alzheimer's Disease Family Study: Genome-Wide Association Study for Susceptibility Loci". The genotypic and associated phenotypic data used in the study, "Multi-Site Collaborative Study for Genotype-Phenotype Associations in Alzheimer's Disease (GenADA)" were provided by the GlaxoSmithKline, R&D Limited. The datasets used for analyses described in this manuscript were obtained from dbGaP at https://www.ncbi.nlm.nih.gov/gap through dbGaP accession number phs000219.v1.p1. Funding support for the "Genetic Consortium for Late Onset Alzheimer's Disease" was provided through the Division of Neuroscience, NIA. The Genetic Consortium for Late Onset Alzheimer's Disease includes a genome-wide association study funded as part of the Division of Neuroscience, NIA. Assistance with phenotype harmonization and genotype cleaning, as well as with general study coordination, was provided by Genetic Consortium for Late Onset Alzheimer's Disease. The datasets used for analyses described in this manuscript were obtained from dbGaP at https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000168.v1.p1.
No founding source is given for the present paper.
The authors declare that they have no conflict of interest.