Skin manifestations of diabetes mellitus (DM) occur as a result of impaired insulin signalling, insulin resistance and chronic hyperglycemia. The biophysical properties of the outer barrier of the epidermis the stratum corneum (SC), reflect the status of both epidermal function and systemic conditions, with over two-thirds of DM patients known to experience skin involvement.
It is accepted that changes to the skin barrier differ between type-1 (T1DM) and type-2 diabetes mellitus (T2DM) patients, but the underlying pathologies driving these disparities remain poorly characterized. In both human and mouse T1DM and T2DM models, complications arise from changes in skin barrier permeability, delayed permeability barrier recovery, impaired SC hydration, altered lipid and glycan homeostasis and impaired keratinocyte proliferation and differentiation. Differences in some of these processes have been documented between T1DM and T2DM cases and likely reflect the specific skin manifestations associated with each DM-type.
In this review, we discuss the current knowledge regarding skin impairment in patients with T1DM and T2DM derived from both human and animal studies. We compare the pathological mechanisms of skin manifestations in each DM-type at the molecular and cellular level, and their associated interplay of etiological factors. How raised blood glucose cannot solely explain the skin manifestations observed in DM is highlighted. We use this information to reaffirm the importance of a dermatologist at the early stages of both T1DM and T2DM diagnosis.
Diabetes mellitus, Skin, Stratum corneum, Pathology, Epidermal permeability, Barrier function
DM is now a worldwide epidemic that continues to increase due to changes in lifestyle and increasing obesity. The prevalence of DM is 415 million people in the world as of 2022, estimated as 10% of the world's adult population. This figure is expected to rise above 600 million by 2040 [1]. The likelihood of DM increases with age, with ~25% of the population above 65 years of age diagnosed [2]. DM is broadly classified into type 1 diabetes (T1DM), type 2 diabetes (T2DM), and gestational diabetes. Other less common types include monogenic diabetes and secondary diabetes. The majority of DM cases are T2DM (~90%), which is more common in males and those aged ≥ 60 years [3]. Chronic hyperglycemia in combination with other metabolic aberrations in DM patients can lead to life-threatening health complications, including microvascular (retinopathy, nephropathy, and neuropathy) and macrovascular complications, which increase the risk of cardiovascular disease [4,5]. T1DM occurs as a result of the autoimmune destruction of pancreatic β cells leading to an absolute deficiency of insulin. A combination of genetic and environmental factors have been implicated as triggers for this autoimmunity [5]. T1DM is most common in children and adolescents, though it can develop at any age. T2DM develops due to insulin resistance typically as a result of obesity and a deficiency in insulin secretion [6]. T2DM is generally lifestyle-related, though genetic predisposition has been reported in patients with neonatal diabetes [7]. In both DM-types, insulin and its analogues can prevent hyperglycemia and protect against long-term DM-mediated complications [8,9].
A variety of dermatologic manifestations have been linked with DM. These skin changes offer insight into the glycemic status of patients and often represent the first signs of metabolic abnormalities [10]. Changes in the biophysical properties of the skin have been reported in both T1DM and T2DM human and animal studies, but the differences between each DM-type remain poorly defined [11]. Recognition and management of these manifestations is important in maximizing the quality of life of patients and for the avoidance of serious adverse effects. Herein, we summarize these epidermal alterations and their implications for the management of DM.
The epidermis acts as a physical barrier to the external environment, preventing water loss from the skin and providing protection to external insults and pathogenic microorganisms. Numerous factors regulate this barrier, including corneocytes, lipids, junction proteins, proteases and antimicrobial peptides [12]. Keratinocytes play an essential role in the formation and maintenance of the epidermal barrier [13]. These cells traverse all layers of the epidermis through proliferation and differentiation, the perturbation of which can impair the skin barrier [14]. Increased permeability of the barrier allows exogenous substances and microorganisms to penetrate the body, promoting a series of adverse immunological reactions [15,16].
The epidermis is composed of five layers: (i) The stratum basale that forms the deepest layer of epidermis consisting of cuboidal and columnar cells; (ii) The stratum spinosum that is composed of skin cells connected by desmosomes that show a spiny appearance upon imaging under a light microscope; (iii) The stratum granulosum that consists of skin cells with granules that contribute to the formation of the outer skin layer; (iv) The stratum lucidum, a thin light layer; (v) And the stratum corneum (SC), the outermost skin layer that serves as the major barrier to microbial, chemical, physical and mechanical insults (Figure 1), [17-22]. The barrier properties of the SC are dependent upon its 10 to 20 layers of flattened corneocytes, divided into the stratum compactum and stratum disjunctum [23-27]. The stratum compactum forms a dense and cohesive layer to which the stratum disjunctum lies superficially (Figure 1). Structurally, the SC is referred to as “bricks and mortar”, in which corneocytes are the bricks and lipids are the mortar [24,28]. Corneocytes consist of water and microfibrillar keratin surrounded by a cornified envelope consisting of densely crosslinked layers of filaggrin, loricrin and involucrin [29]. A monolayer of non-polar lipids is esterified to the cornified envelope, which forms a template for intercellular lipid layers [30]. These with the cornified envelope, minimize the non-specific uptake of substances into the corneocytes, permitting the correct formation of the lipid matrix [31,32]. Recent 3D time-of-flight secondary ion mass spectrometry analysis revealed a distinct distribution of cholesteryl oleate independent from the lipid matrix separating the corneocyte layers [20,33]. Chronic hyperglycemia, oxidative stress and altered lipid content promote deterioration of the SC in DM animal models due to changes in hydration status, lipid composition and glycan levels in the barrier [34-37].
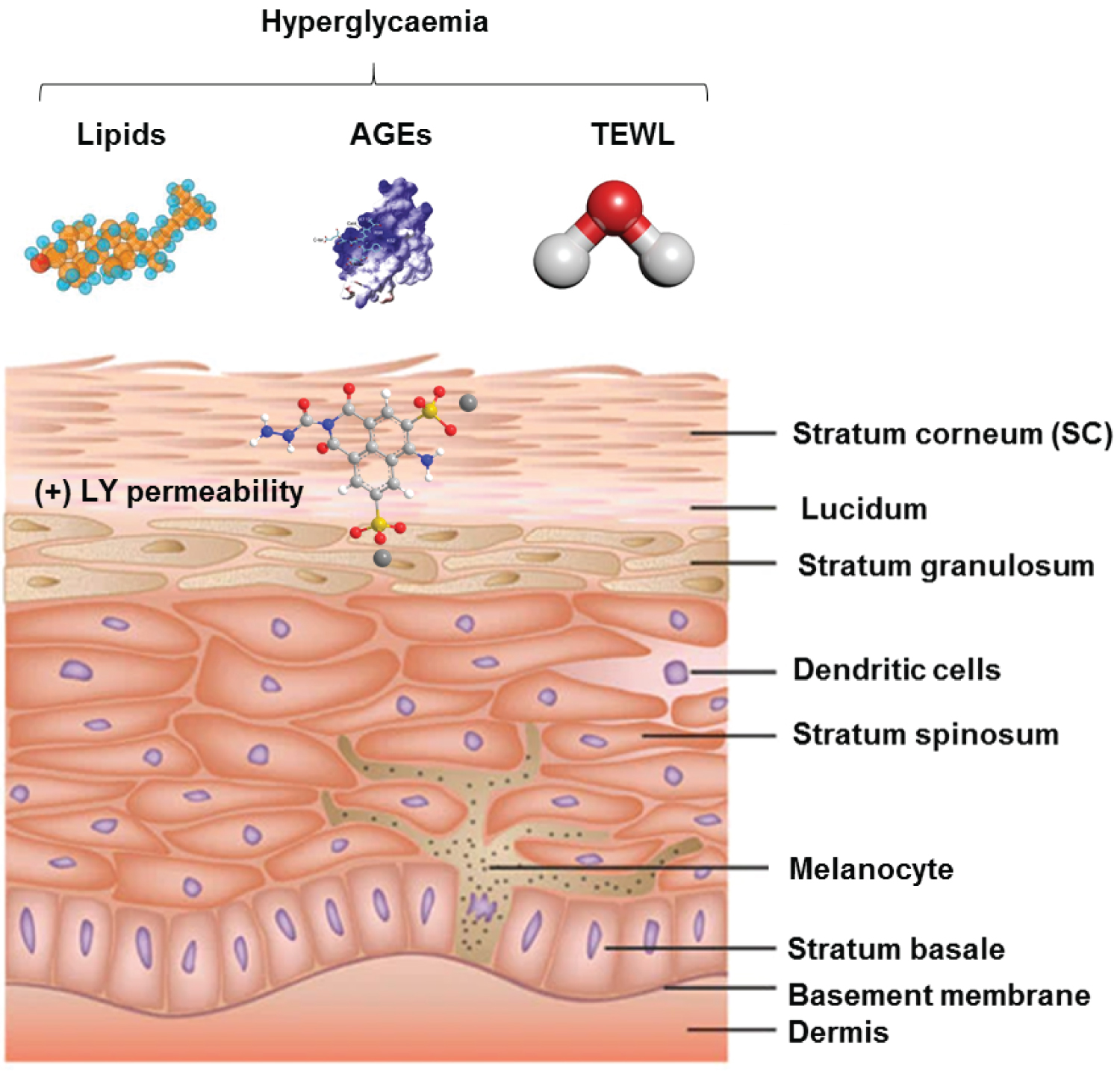 Figure 1: Structure and regulation of the epidermal barrier. The epidermis is composed of five layers: (i) The stratum basale that forms the deepest layer of epidermis consisting of cuboidal and columnar cells; (ii) The stratum spinosum that is composed of skin cells connected by desmosomes that show a spiny appearance upon imaging under a light microscope; (iii) The stratum granulosum that consists of skin cells with granules that contribute to the formation of the outer skin layer; (iv) The stratum lucidum, a thin light layer; (v) And the stratum corneum (SC), the outermost skin layer that serves as the major barrier to external insults (microbial, chemical, physical and mechanical). The biophysical properties of the outer barrier of the epidermis, the stratum corneum can be altered during DM due to complications arising from changes in SC hydration, altered lipid homeostasis, increased permeability and delayed permeability barrier recovery. This increases accessibility for the normally non-permeant lucifer yellow (LY) dye.
View Figure 1
Figure 1: Structure and regulation of the epidermal barrier. The epidermis is composed of five layers: (i) The stratum basale that forms the deepest layer of epidermis consisting of cuboidal and columnar cells; (ii) The stratum spinosum that is composed of skin cells connected by desmosomes that show a spiny appearance upon imaging under a light microscope; (iii) The stratum granulosum that consists of skin cells with granules that contribute to the formation of the outer skin layer; (iv) The stratum lucidum, a thin light layer; (v) And the stratum corneum (SC), the outermost skin layer that serves as the major barrier to external insults (microbial, chemical, physical and mechanical). The biophysical properties of the outer barrier of the epidermis, the stratum corneum can be altered during DM due to complications arising from changes in SC hydration, altered lipid homeostasis, increased permeability and delayed permeability barrier recovery. This increases accessibility for the normally non-permeant lucifer yellow (LY) dye.
View Figure 1
Regarding skin pH (typically 4.1-5.8 in healthy subjects), more acidic values have been recorded in T1DM patients compared to control subjects [38,39]. Differential SC water homeostasis between T1DM and T2DM have been reported and may contribute to differences to the differences in skin integrity observed between these DM types. Sakai, et al. [40,41] reported a reduced hydration state combined with decreased sebaceous gland activity in T1DM mice administered either STZ or alloxan [42]. Later studies by Lehman, et al. [43] assayed alterations in SC barrier function through assessment of the percutaneous absorption of water, ethanol, lidocaine and hydrocortisone in 4-week T1DM and 8-day insulin-treated T1DM Sprague-Dawley CD rats [43]. They observed subtle changes in the total penetration and peak flux of lidocaine and hydrocortisone in T1DM compared to control groups, but significantly increased ethanol flux indicative of altered SC physiology [44-46]. Importantly these alterations were improved by insulin therapy [43].
In human studies, Seirafi, et al. reported no significant changes in TEWL in 34 T2DM patients [47], which was disputed by later studies documenting a significantly reduced TEWL compared to controls in a larger cohort of T2DM subjects [48]. In T2DM models however, increases in skin pH (5.95 ± 0.02 vs. 5.68 ± 0.04) are common [38,49]. This concept aligns with the increased susceptibility of T2DM vs. T1DM patients to skin infections due to the known protective of an acidic skin barrier to microbial infections [50]. Differences in skin dryness have been documented in T1DM ST induced-mice, non-obesity T2DM model mice and obesity T2DM DM mouse models [51]. Increases in trans epidermal water loss (TEWL) were also observed in both T1DM and T2DM mice [40,41,51].
Collectively, these data highlight key differences in barrier function between T1DM and T2DM. Current consensus suggests that SC barrier function is impaired in T1DM, but the initial compensatory hyperinsulinemia observed in T2DM can reduce glucose levels during early disease phases to sufficiently minimize the impact of hyperglycemia on skin barrier integrity [52,53].
SC barrier function remains poorly defined in DM patients, but altered states of SC hydration, pH and lipid content have been reported. Okano, et al. [35] investigated the effects of T1DM on the barrier in streptozotocin (STZ) induced mouse models [54-57], using lucifer yellow (LY) staining (diameter of ~0.95 nm) due to its impermeability to an intact epidermis [58]. In control mice, the dye could not penetrate the skin, contrasting the high levels of staining observed during insulin deficiency in diabetic mice. Insulin injections in T1DM mice restored LY staining to control levels thereby restoring barrier integrity. This highlighted hyperglycemia as a major driver of SC barrier dysfunction during T1DM [35]. These contrasted studies performed in T2DM mouse models that showed little-to-no impairment in SC integrity compared to control mice as assessed by hematoxylin and eosin (H&E) staining [36].
Serum lipid abnormalities are present in ~75% of DM patients predisposing them to a range of lipid-associated pathologies [59]. Skin surface lipids are derived from the epidermis and sebaceous glands, whilst intercellular lipids of the SC are supplied by sebum that is produced in the sebaceous glands and is rich in triglycerides, wax esters and squalene [60]. The correct composition of lipids within the SC is critical to epidermal homeostasis.
Changes have been documented in both T1DM and T2DM cases, the lipid profiles of which are under exploration for potential biomarkers to expedite early disease diagnosis [61]. As an example, high levels of skin-derived free fatty acids (FFAs) and triglycerides have been documented in both T1DM and T1DM models [62-64]. In the SC, the most abundant short-chain FFAs are C16: 0 and C18: 0. The most abundant long-chain FFAs are C24: 0 and C26: 0 [65]. The levels of C15, C17, C18: 1 and C23 in the SC significantly increase in young diabetic subjects [66]. Such increased levels of FFAs and their composition have been linked to a range of skin diseases including xerosis and scleroderma, both of which are prevalent in T1DM and T2DM patients [66,67].
The SC also comprises an abundance of cross-linked proteins, including transmembrane glycoproteins and proteoglycans covalently bound to a monolayer of cell surface ceramides. Both the total levels of glycans and their specific distribution are thought to contribute to the progressive changes of the SC, as it evolves from the SC compactum to the SC disjunctum, towards desquamation [68]. Hyperglycemia in DM patients increases plasma protein binding to collagen through glycation [69-71]. High levels of Advanced-Glycation-End (AGE)-products [72], are not restricted to DM, and can occur in cases of renal insufficiency, smoking and as a result of a modern western diet consisting of high levels of processed foods [73-75]. A glycated epidermis reduces the elasticity of the skin barrier [71]. Niu, et al. analyzed skin biopsies of 19 T2DM patients (mean age 59.0 ± 8.2 years, mean HbA1c 8.4% ± 0.6%) and 14 age matched controls and observed that the content of AGE T2DM skin was significantly higher than control samples [70]. Of note, these samples were collected from a burns unit, with patients suffering from stress-induced hyperglycemia [76] and require validation in a larger cohort.
AGEs accumulate in long-lived proteins such as fibrillary collagen, resulting in structural alterations to the skin [69,71,77]. AGE compounds such as N-epsilon-carboxy-methyl-lysine (CML), carboxy-ethyl-lysine (CEL), 5-hydro-5-methylimidazolone (MG-H1) are expressed in tissue and blood plasma [69] and bind to AGE receptors (RAGE), resulting in vascular complications in DM patients [72,78]. Binding between AGEs and RAGE also promotes oxidative stress and inflammatory cascades via the activation of mitogen- activated protein kinases (MAPK), nuclear factor-k-light-chain-enhancer of activated beta cells (NF-kbeta), interleukin- 6 (IL-6), and tumor necrosis factor-alpha (TNF-alpha) [79]. A systematic review by Putte, et al. analyzed fourteen articles to dissect the influence of AGEs in DM skin [80] in which AGE-accumulation was shown to promote the formation of disorganized, shortened and thinned collagen, reducing skin elasticity and wound healing. This parallels the skin changes observed in DM, highlighting the likely contribution of AGEs to the DM-induced loss of skin elasticity [81].
Building on these studies, Peeters, et al. performed a cross-sectional analysis of the association between AGEs and matrix metalloproteinases (MMPs) in 670 T1DM patients [82]. They identified an independent association of circulating serum AGEs, (CML, CEL and H1MG) with MMPs (MMP-2, MMP-3) and tissue inhibitor of metalloproteinase (TIMP-1) expression [82] in which serum levels of protein-bound AGEs were measured using liquid chromatography and concentrations of MMP-TIMP measured using ELISA [82]. The observed changes in MMP-TIMP expression resulted in pathological changes including the enhanced production of proinflammatory cytokines and altered extracellular matrix (ECM) composition [82]. Consequently, this leads to reduced collagen turnover in the ECM, leading to a thinner and thus more fragile dermis in T1DM subjects [70].
A number of questions from these studies remain. As AGEs are not exclusive to DM, other risk factors may be involved in their production. In T1DM, it is unclear if AGE production is causative or occurs as a result of T1DM development.
In T2DM, comparison between newly diagnosed and long-standing T2DM cases would provide data on the contribution of AGEs to disease progression. If proven as a causative factor, early intervention with targeted anti-AGE therapy may prevent such deleterious effects. A complete temporal analysis of this association is therefore required in future studies.
In both T1DM and T2DM, a high glucose environment prevents the normal proliferation of skin cells required for a healthy SC [71,83,84]. Keratinocytes constitute the major cell type of the epidermis and participate in re-epithelialization during wound repair and immune defense responses to pathogen invasion (Figure 2). Altered skin wound healing is common amongst DM patients, with hyperglycemia and impaired insulin signalling known to suppress the proliferation and differentiation of both human and rodent keratinocytes [83,85]. Insulin itself can promote wound healing through its ability to enhance the release of Vascular Endothelial Growth Factor (VEGF) from keratinocytes in skin wounds, through Akt1-mediated posttranscriptional mechanisms [86]. Keratinocyte Growth Factor (KGF) release has been shown to be suppressed in T2DM Adipose-derived stem cells (ASCs) compared to non-diabetic ASC groups, likely contributing to the reduction in wound healing observed in T2DM cases [87].
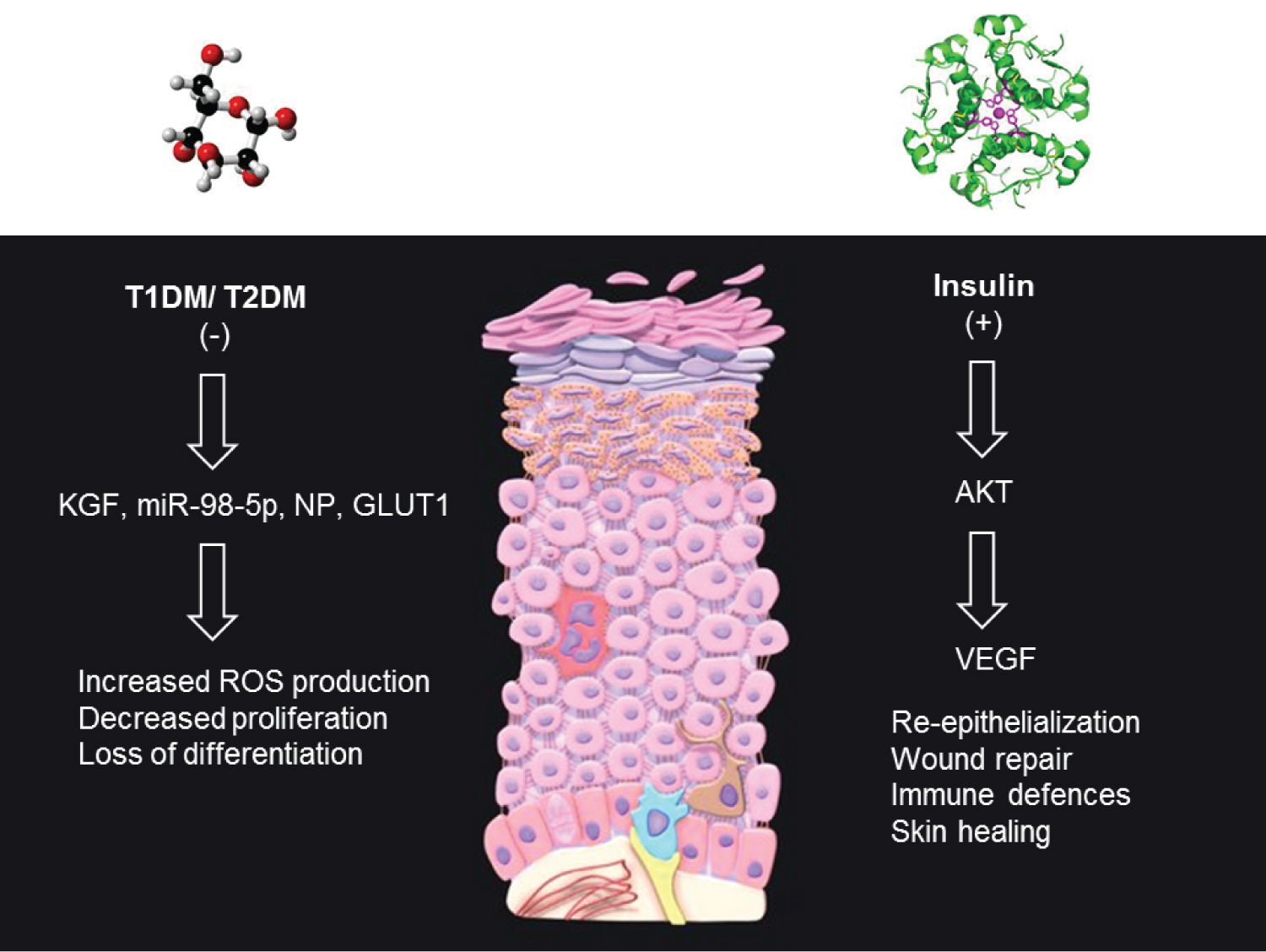 Figure 2: Altered keratinocyte functions in T1DM and T2DM. Keratinocytes represent the major cell type of the epidermis and participate in re-epithelialization during wound repair and immune defenses. In both T1DM and T2DM, hyperglycemia prevents the normal proliferation of skin cells required for a healthy SC through effects of KGF, MiRs, NP and GLUT1 expression. These effects are attributed to the suppression of insulin secretion and/or insulin sensitivity. Insulin can promote wound healing through its ability to enhance the release of Vascular Endothelial Growth Factor (VEGF) from keratinocytes in skin wounds, through Akt1-mediated posttranscriptional mechanisms.
View Figure 2
Figure 2: Altered keratinocyte functions in T1DM and T2DM. Keratinocytes represent the major cell type of the epidermis and participate in re-epithelialization during wound repair and immune defenses. In both T1DM and T2DM, hyperglycemia prevents the normal proliferation of skin cells required for a healthy SC through effects of KGF, MiRs, NP and GLUT1 expression. These effects are attributed to the suppression of insulin secretion and/or insulin sensitivity. Insulin can promote wound healing through its ability to enhance the release of Vascular Endothelial Growth Factor (VEGF) from keratinocytes in skin wounds, through Akt1-mediated posttranscriptional mechanisms.
View Figure 2
Using next-generation sequencing (NGS), an array of genes (~420) have been identified as differentially expressed in DM vs. non-DM keratinocytes, providing new clues into understanding the pathogenesis of epidermal pathologies in DM patients [88]. The transcriptional characteristics of keratinocytes at the single-cell level from T2DM patients were also recently scrutinized using single-cell RNA sequencing from skin tissues of two T2DM patients. A total of 356 differential genes were identified, with enriched pathways including oxidative phosphorylation, antigen processing, bacterial invasion of epithelial cells, vasopressin-regulated water reabsorption, and protein processing in the endoplasmic reticulum amongst others [89]. Deregulated circulatory microRNA (miRNA) signatures during DM have been extensively identified but only recently linked to the progression of impaired skin health during DM. Khan, et al. identified miR-98-5p as downregulated in DM keratinocytes which leads to increased apoptosis and decreased proliferation through targeting PPP1R15B [90]. Impaired wound healing in DM patients also leads to the development of chronic wounds that manifest in the feet as a result of neuropathy and/or vascular disease [91]. Diabetic foot ulcers (DFUs) have also been linked to impaired wound healing involving endothelial cells, immune cells, keratinocytes and fibroblasts. Recent studies identified that DM patients with healing DFUs possess a unique population of fibroblasts that overexpress MMP1, MMP3, MMP11, HIF1A, CHI3L1, and TNFAIP6 and a higher abundance of M1 macrophages compared to non-healing DFUs [91]. Neuropeptides such as substance P (SP) play an important role in wound healing by promoting neovascularization, inhibiting proinflammatory chemokine expression and regulating macrophage polarization. SP and CGRP have shown to be downregulated in DM and DFUs [92].
Skin constitutes approximately 15% of the total body weight and reduced glucose uptake by keratinocytes can reciprocally drive hyperglycemia [83,88], This loss of glucose uptake occurs as a result of decreased GLUT1 expression in proliferating keratinocytes exposed to high glucose in DM cases [83]. Glucose exposure enhances the production of reactive oxygen species (ROS), augments inflammatory responses and reduces antioxidant function [93,94]. Increased ROS production can further delay wound healing and promote retinopathy, neuropathy, and nephropathy, all known symptoms of DM [95,96]. Shen, et al. characterized the complement of GLUT receptors in skin keratinocytes [96], showing that Ca 2+ exposure increases the expression of GLUT 1, 2 ,3, and 5; all of which mediate glucose transport. Zhang, et al. later confirmed high GLUT1 expression in the proliferative basal layer of the epidermis in both DM human and mouse skin [84]. Silencing of GLUT 1 in mouse models led to attenuated keratinocyte proliferation and increased oxidative stress, also frequently observed in DM skin samples [34,84].
A relationship between insulin signalling and skin barrier function has also been proposed [96-100]. For optimal skin barrier function, epidermal stem cells must proliferate and differentiate on-cue [99] and insulin receptors (IR) are highly expressed in keratinocytes [100,101]. Muraguchi, et al. further explored the links between IGF/IGF-1R signalling and epidermal homeostasis in IGF-1R knockout (KO -/-) human keratinocytes [99]. The addition of IGF-1 into the culture media enhanced the proliferation of keratinocytes, which contrasted IGF-1R -/- cells that displayed epidermal hypoplasia.
These findings contrasted those of Okano, et al. who suggested that in diabetic mouse models, epidermal disruption occurs as a result of impaired keratinocyte proliferation due to hyperglycemia as opposed to impaired insulin signalling [19]. It is likely that the interplay between hyperglycemia and impaired insulin signalling collectively impair glucose utilization by skin keratinocytes resulting in the loss of skin proliferation and differentiation required for homeostasis of the SC barrier [35,36,84,100-102].
Insulin treatment activates Akt to maintain glucose homeostasis [102]. Yu, et al. evaluated the role of insulin in STZ-induced diabetic rats fed a high fat diet (HFD) [100], revealing low expression of IR, IGF-1, GLUT-1, and phospho-AKT in diabetic rats prior to insulin injection compared to controls [100]. Following insulin treatment, increased levels of phospho-AKT and high insulin sensitivity in diabetic skin tissue were observed compared to the liver tissues of DM mice [100]. Furthermore, increased translocation of GLUT1 from the cytosol to the surface of basal epidermal cells was observed in diabetic rats in response to insulin.
Skin problems represent an early visible sign of DM, the progression of which leads to new skin disease and enhances the pathology of existing skin problems. These changes induced by hyperglycemia negatively impact skin integrity, enhance susceptibility to infection/insults and suppress wound healing. Skin manifestations are frequent in both T1DM and T2DM, with the prevalence of specific DM-associated skin diseases more common in each DM type. These are likely due to differences in the underlying pathophysiology of T1DM vs. T1DM (suppression of insulin secretion vs. insulin resistance) and their subsequent impact on SC determinants such as lipids, glycans and water content.
To advance this field, and achieve a more complete understanding of the skin-associated changes in both T1DM and T2DM, and how they contrast and differ in controlled studies must be performed to produce a consistent narrative of tractable skin alterations. Identification of the associated genes and pathways affected can also open up new horizons into early and personalized therapeutic targets to treat skin disease in both DM-types. Studies using NGS and single-cell RNA sequencing have been used to identify changes in skin cells at the gene expression level in T2DM. These studies are in their infancy and now require extrapolation into larger patient cohorts of both T1DM and T1DM patients to identify genes and pathways associated with each skin phenotype and/or disease.