Hepatosplenic T-cell lymphoma (HSGDTL) is a rare variant of T-cell lymphoma with few reported cases in literature. It usually presents with hepatosplenomegaly and constitutional symptoms, primarily in young to middle aged patients. The prognosis is generally poor, with maximum reported survival periods of 2-3 years. There is a link to immunodeficiency and some biological treatments. Liver biopsy with flow cytometry is required to confirm the diagnosis.
We report a case of gamma delta (γδ) HSTCL in a 37-year-old female presenting as progressive hepatosplenomegaly with cytopenia during pregnancy. The patient underwent chemotherapy, splenectomy and bone marrow transplant and is in remission 10 months after the diagnosis.
As far as we are aware, this is the first case of γδ HSGDTL reported in the Middle East and the second documented case of HSGDTL that highlighted during pregnancy.
Hepatosplenic T-cell lymphoma, Gamma delta T-cell lymphoma, Pregnancy
Hepatosplenic gamma delta (γδ) T-cell lymphoma (HSGDTL) is a rarely encountered peripheral T-cell lymphoma. It accounts for less than 1 percent of all Non-Hodgkin Lymphomas [1]. Prognosis is poor, although data is limited because of its relative rarity. Patients are usually young to middle aged males, 30-40 years-old. Less than 20 percent of cases show possible link to immunosuppression such as organ transplant and inflammatory bowel disease patients exposed to immunomodulators [2].
The underlying mechanism of this disorder is unknown, although association with isochromosome 7q has been reported [3]. Most HSTCL have the gamma-delta subtype of the T-cell receptors (TCR) with few cases reported to have the alpha-beta TCR subtype [4].
As the name suggests, HSGDTL presents with hepatosplenic involvement, causing massive hepatosplenomegaly. Lymphadenopathy is not common. Patients have pancytopenia or at times, only thrombocytopenia. Constitutional symptoms are varied and most cases are also reported to have bone marrow involvement.
Chemotherapy is the mainstay of treatment, but the outcome is generally poor. Splenectomy has been reported to be beneficial in patients with thrombocytopenia, prior to the start of chemotherapy [5].
To our knowledge, only one report has documented the development of HSGDTL during pregnancy. This was diagnosed post-splenectomy with remission achieved after chemotherapy [6]. Another case was reported to have developed the disease during the post-partum period, with an otherwise healthy and uneventful pregnancy [7].
Herein, we describe a case of HSGDTL, presenting with progressive marked hepatosplenomegaly and pancytopenia during early pregnancy, the splenomegaly was noted just before pregnancy initiation as the patient presented with vague abdominal pain and constipation, during pregnancy hepatosplenomegaly and pancytopenia became more evident and diagnosis was confirmed with liver biopsy after delivery.
A 37-year-old Egyptian female with no significant medical history presented with vague abdominal pain and constipation, 1 month before pregnancy. Ultrasound revealed spleen size of 16.3 cm with prominent splenic vein and some collaterals at left splenic hilum, and a subsequent colonoscopy was normal and no action was taken at that time.
At 8 weeks gestational age, the patient presented again with left sided abdominal pain and heaviness with radiation to the left shoulder, fatigue and frequent fever. Her platelet and white blood cell (WBC) counts were low (98000/µL and 2100/µL respectively) and haemoglobin 11 gm/dL. Abdominal ultrasound revealed hepatosplenomegaly with spleen measuring 21 cm and liver measuring 16.6 cm.
Autoimmune panel and viral serology were negative. No cause for the hepatosplenomegaly was identified. The patient was put on conservative management with repeated ultrasound showing that the hepatosplenomegaly was progressive and reached massive splenomegaly at the third trimester.
The patient underwent elective lower segment caesarean section at 33 weeks, delivering a healthy child.
Post-nataly the patient presented to our hospital with continuing symptoms of pallor, tiredness.
Physical examination revealed pallor and protruded abdomen with tender splenomegaly exceeding the umbilicus, liver span of 18 cm, no chronic liver disease stigmata, and no enlarged lymph nodes.
Laboratory work up showed pancytopenia with WBCs of 1200/µL, Hb of 7.2 gm/dl and platelets of 74000/µL with normal kidney functions, ALT 14 unit/L, AST 20 unit/L, total bilirubin 14 micromole per litre, albumin 26 gm/L, INR 1.7.
Iron saturation 26%, normal Hb electrophoresis, ESR 20 mm/hr, LDH 92 unit/L.
Ultrasound of the abdomen revealed progressive hepatosplenomegaly (spleen measuring 33 cm) with collaterals around the splenic hilum and small amount of free fluid. Whole body PET CT scan revealed further increasing size of spleen and liver, mild peripheral splenic uptake with indeterminate etiology and no lymphadenopathy (Figure 1).
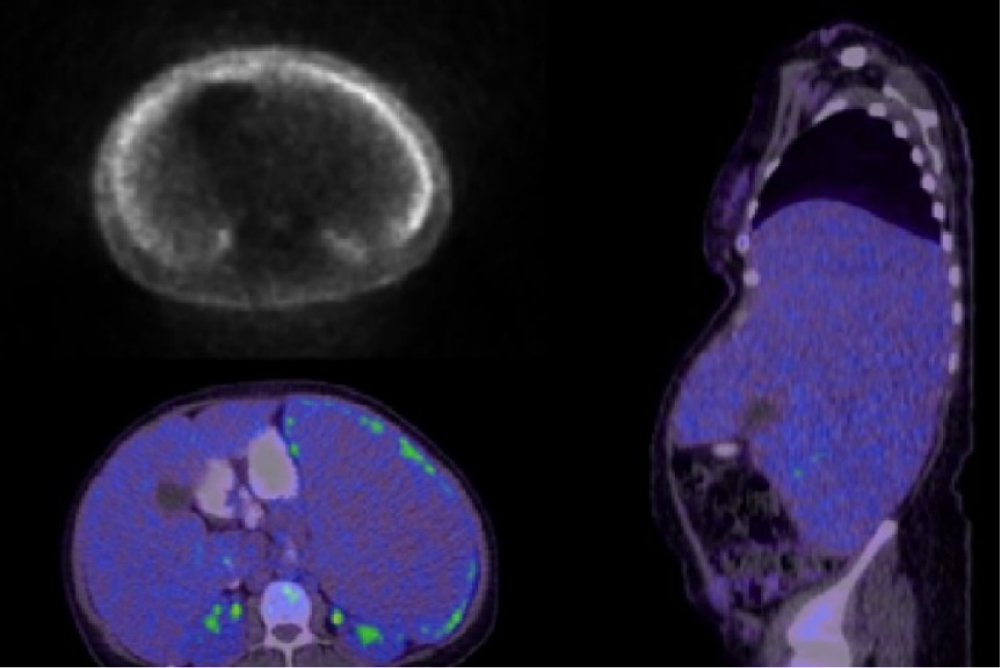 Figure 1: PET CT scan showing hepatosplenomegaly with mild peripheral splenic uptake of indeterminate aetiology, no lymphadenopathy.
View Figure 1
Figure 1: PET CT scan showing hepatosplenomegaly with mild peripheral splenic uptake of indeterminate aetiology, no lymphadenopathy.
View Figure 1
Hepatitis serology, HIV, malaria smear, (autoimmune hepatitis, leishmaniasis, Wilson's disease) work up, Schistosoma antibodies, EBV & CMV PCR were all negative.
Peripheral smear showed red cells were normochromic with mild anisocytosis, few oval and tear drop cells with increased rouleaux and rare nucleated red blood cells. There was leukopenia and severe neutropenia, majority of the lymphocytes seen were small mature with rare showing irregular nuclear contour. Platelets were markedly reduced in number, cells differential: Neutrophils: 57%, Lymphocytes: 26%, Monocytes: 16%, Eosinophils: 1%.
Liver biopsy was subsequently performed. The biopsy showed expansion of the hepatic sinusoids by monomorphic infiltrate of small lymphoid cells (Figure 2 and Figure 3). No periportal infiltrates were seen. The intrasinusoidal lymphoid cells were positive for CD2, CD3 (Figure 4) and CD7, confirming the cells to be of T cell origin. Gamma delta immunohistochemistry was also positive (TCR delta partial, weak positive). The cells also co-expressed CD56 (Figure 5) and had a non-activated cytotoxic (TIA-1 positive, granzyme B negative) lymphocytic phenotype. Other markers including CD20, CD5, CD4 and CD8 were negative. The immunophenotypic data, in conjunction with the morphology, supported a diagnosis of γδ HSGDTL. FISH analysis did not show isochromosome 7q or trisomy 8. Bone marrow involvement was also confirmed on biopsy.
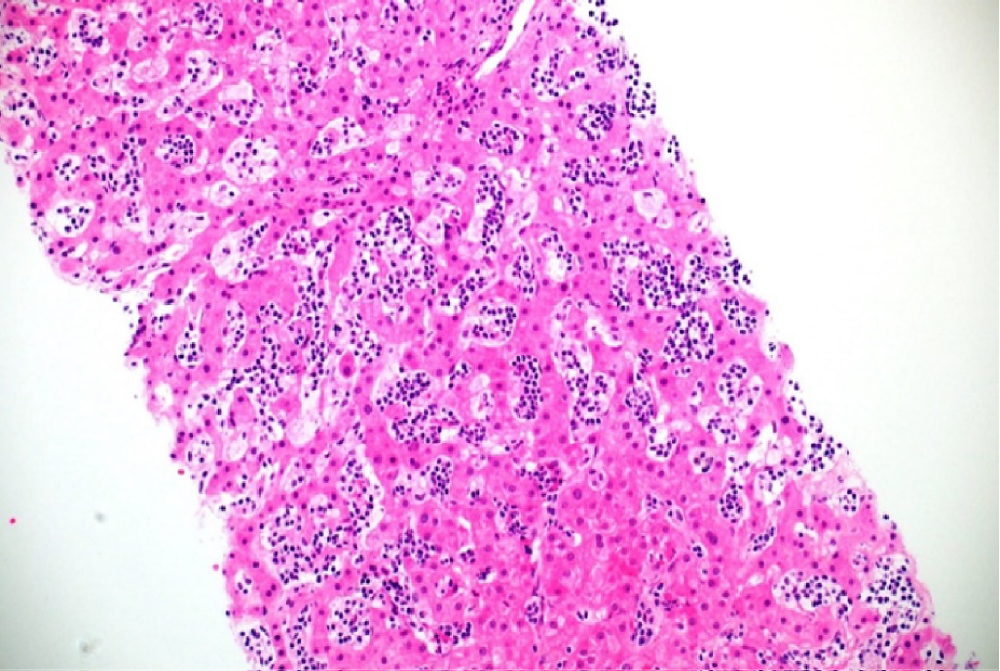 Figure 2: Expansion of the sinusoids by monomorphic infiltrate of small lymphoid cells. Haematoxylin and Eosin X20.
View Figure 2
Figure 2: Expansion of the sinusoids by monomorphic infiltrate of small lymphoid cells. Haematoxylin and Eosin X20.
View Figure 2
 Figure 3: Higher power view showing sinusoidal infiltration of small lymphoid cells. Haematoxylin and Eosin X40.
View Figure 3
Figure 3: Higher power view showing sinusoidal infiltration of small lymphoid cells. Haematoxylin and Eosin X40.
View Figure 3
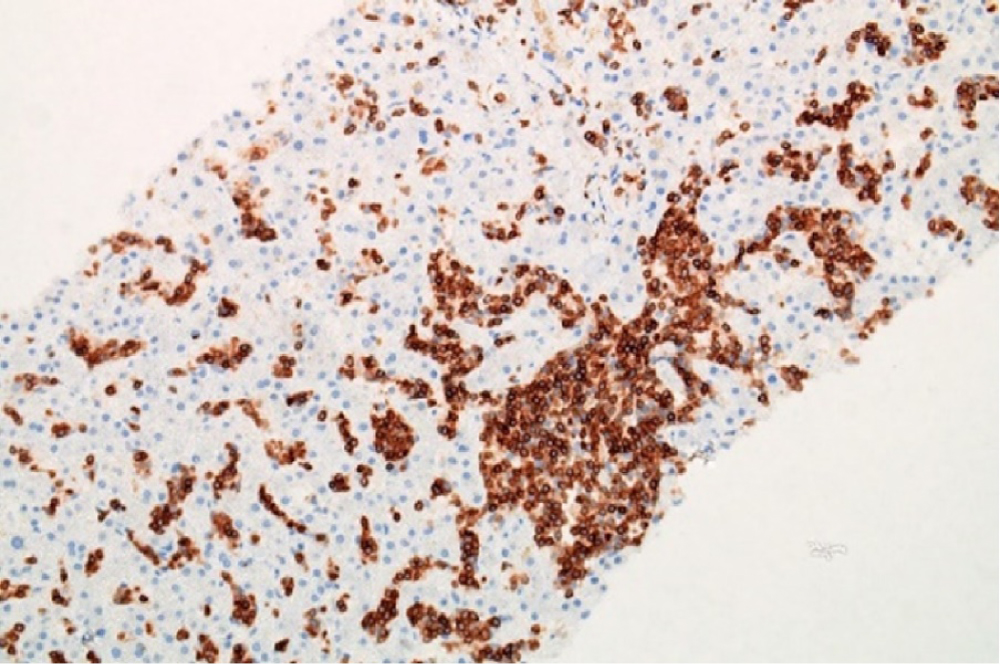 Figure 4: The lymphoid cells are positive for T cell marker, CD3.
View Figure 4
Figure 4: The lymphoid cells are positive for T cell marker, CD3.
View Figure 4
 Figure 5: The T cells are also CD56 positive.
View Figure 5
Figure 5: The T cells are also CD56 positive.
View Figure 5
Following the diagnosis, the patient received Cyclophosphamide, Vincristine, Prednisolone (COP) and dexamethasone, (1 cycle) followed by two cycles of Ifosfamide, Carboplatin and Etoposide (ICE) chemotherapy regimens. The general condition of the patient improved and her blood counts steadily increased. Five months after the initial diagnosis, splenectomy was performed, histology of which also showed involvement by γδ HSGDTL. An allogenic stem cell transplant from Human Leukocyte Antigen (HLA) matched sibling donor was subsequently undertaken. 10 months post-partum, the patient is in clinical remission, with no evidence of disease radiologically or in her bone marrow.
The baby was doing fine at that point.
HSGDTL is a rare Non-Hodgkin's Lymphoma, falling under the category of peripheral T-cell lymphomas. T-lymphocytes identify antigens through polypeptides called T cell receptors that are mainly built by alpha (α) and beta (β) chains or rarely γ and δ chains. These receptors undergo rearrangement of their subtypes to produce CD4+/CD8+ state with αβ T cell receptor (TCR) on the surface; and CD4-/CD8- state with γδ TCR on the surface, which in turn are distributed in the lymphoid system with highest concentration in the spleen [2].
The pathogenesis of the transforming events to develop HSGDTL are not well understood, but it was assumed that the triggering event is a monoclonal proliferation with up-regulation of JAK/STAT pathway with or without chromosomal abnormality such as isochromosome 7q trisomy [2].
The majority of HSGDTL express γδ and a minority have αβ TCR, which is now considered as a variant of the same disease entity.
The link to long standing immunosuppression has been reported in less than 20% of the cases of HSGDTL. These cases were either post-transplant patients or were known to have treatment with immunomodulators especially anti-Tumor Necrosis Factor alpha (TNFα), particularly in patients with Crohn's disease and rheumatoid arthritis [8].
The association between pregnancy and the development of lymphoma has been reported previously [8]. Hodgkin's lymphoma has the most prevalent association with pregnancy. Prognosis of these patients was found to be not too dissimilar from non-pregnant cases. Several changes during pregnancy were theoretically linked to the development of lymphoma, including the immune system changes as a result of high levels of progesterone in the blood.
Cytarabine-based chemotherapy was reported to have better survival rates than alkylating agents and anthracyclines, possibly because of significant expression of P-Glycoprotein (encoded by gene MDR-1) and Glutathione-S-transferases [9].
It was suggested also that trial of demethylating agents in this disease because it was found that A1M1 (a gene might have a role in tumor suppression in HSGDTL) is significantly reduced in HSTL most likely due to promoter methylation [9].
It was also suggested that Syk (a protein thyrosin kinase involved in B-cell receptor signalling and activation and it was found to be common in most peripheral T-cell lymphomas) can be an important target for therapy, inhibition of Syk induces apoptosis and blocks proliferation in T-cell lymphoma cell lines [10].
In this report, we wish to highlight a relatively rare lymphoma, hitherto unreported in the Middle East, and presenting during pregnancy as massive hepatosplenomegaly. Recognition of this entity and considering its possibility in the absence of background lymphadenopathy is vital to achieve early diagnosis and treatment.
First three authors contributed equally to this manuscript. Hussam Almasri is the article guarantor.
Nothing to declare.
Patient consent has been obtained for publication of case details.