Background: Acrokeratosis verruciformis of Hopf is a rare genodermatosis with an autosomal dominant mode of inheritance. It is a disorder of keratinization, characterized by multiple, flat-topped, skin-coloured keratotic lesions resembling plane warts typically observed on the dorsum of the hands and feet.
Case presentation: We report a rare sporadic case of a 36-year-old Hindu male presented with multiple, grouped, hyperkeratotic, whitish, flat papules on the dorsum of the palm, which had been present for more than one year. Histopathological examination showed typical findings of acrokeratosis verruciformis of Hopf. Our case is unique because the patient had no familial history of similar skin lesions.
Discussion & conclusion: Histopathologically, these lesions show considerable hyperkeratosis, acanthosis, and papillomatosis, mimicking a "church spire", and a thickened granular layer. It arises in early life, often at birth or infancy.
Non-familial acrokeratosis verruciformis, Hopf disease, Church spire
Acrokeratosis verruciformis (AKV) is a rare hyperkeratotic genodermatosis that was first described by Hopf in 1931 [1]. It typically presents as multiple, small, flat, wart-like papules on the dorsum of the hands and feet and arises in early life often at birth or infancy with no sexual predilection [2,3]. Histologically, it is characterized by hyperkeratosis without parakeratosis, marked papillomatosis without vacuolization, which resembles a “church spire”. It exhibits an autosomal dominant inheritance pattern and shows incomplete penetrance. Because of this, there may not always be a family history. However, a few sporadic cases have also been reported [4]. We herein report on a rare case of AKV, which developed in a young adult without familial history.
We report a rare sporadic case of a 36-year-old Hindu male presented with multiple raised skin-coloured lesions in his hand and trunk which had been present for more than one year. On examination, the lesions were multiple brownish bullous papules to plaque-like grouped, hyperkeratotic, whitish, flat papules on the bilateral dorsum of the hands. The lesions in the trunk were more flat skin coloured to hyper pigmented lesions of 2-5 mm size. Palmer's aspect was spared (Figure 1). There were no complaints of itching, pain or burning sensation. The lesion did not have any sessional variations or any systemic complaints. The Fitzpatrick skin type in our patient was V. Skin sensitivity test for the possibility of allergic dermatitis was done and negative. Clinical differentials were epidermodysplasia verruciformis, Darier's disease, and AKV.
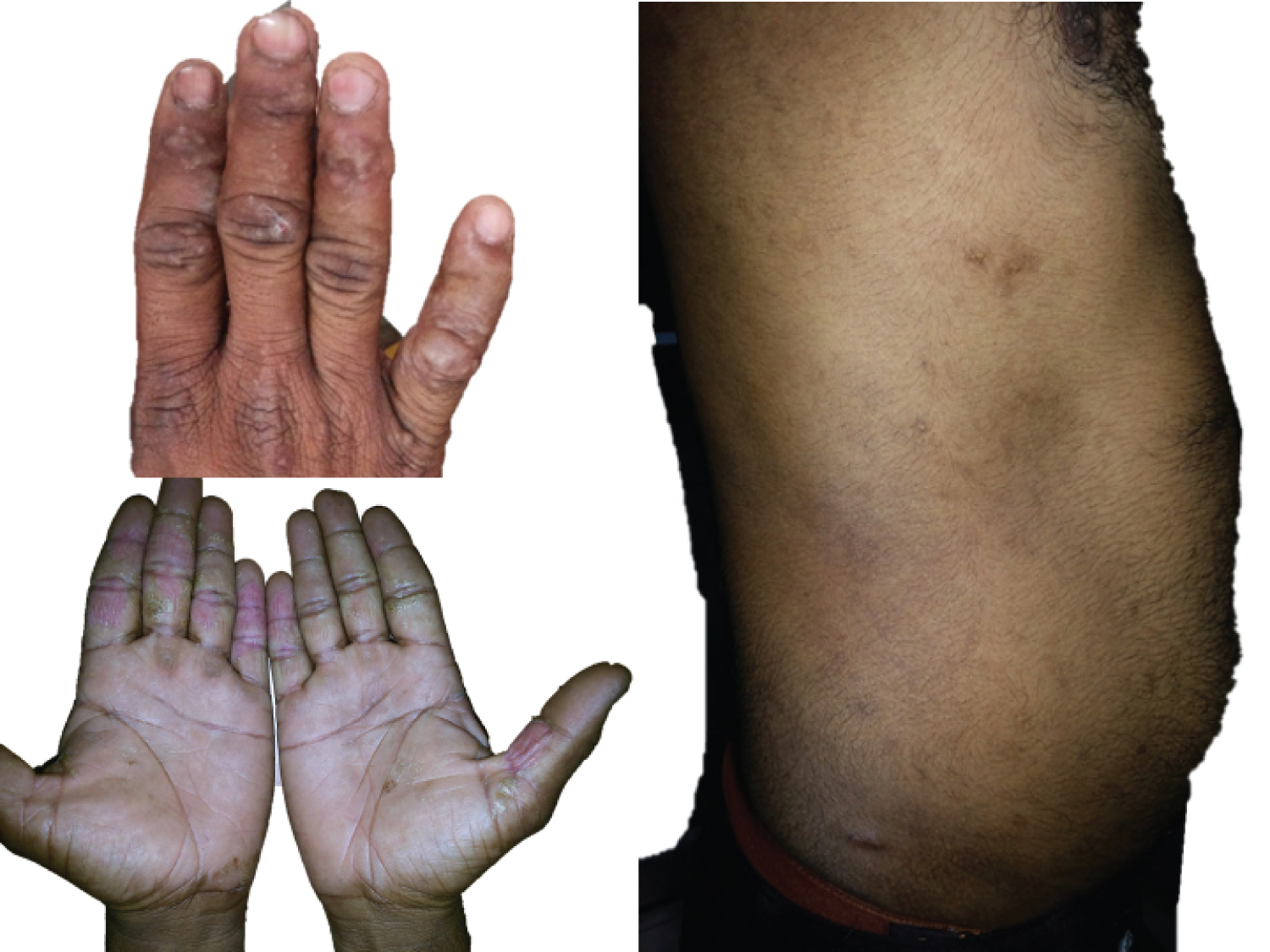 Figure 1: Numerous variably sized flesh hyperkeratotic papules on the dorsal aspect of hand, and trunk along with sparing of palm.
View Figure 1
Figure 1: Numerous variably sized flesh hyperkeratotic papules on the dorsal aspect of hand, and trunk along with sparing of palm.
View Figure 1
Skin punch biopsy from the macule over the left hand was taken. Histopathology showed an epidermis with compact hyperkeratosis, acanthosis, papillomatosis, mild spongiosis, and a thickened granular layer. The typical circumscribed epidermal elevation “Church Spire-like pattern” was noted, with the absence of parakeratosis (Figure 2). No intracytoplasmic inclusions, corps/rounds were noted. The papillary dermis showed mild oedema with sparse perivascular lymphocytic cuffing. The reticular dermis and sub cutis were unremarkable (Figure 3). Based on the clinical and histopathology findings, a diagnosis of Acrokeratosis verruciformis of Hopf. Given financial constraints the patient was not able to undergo genetic testing.
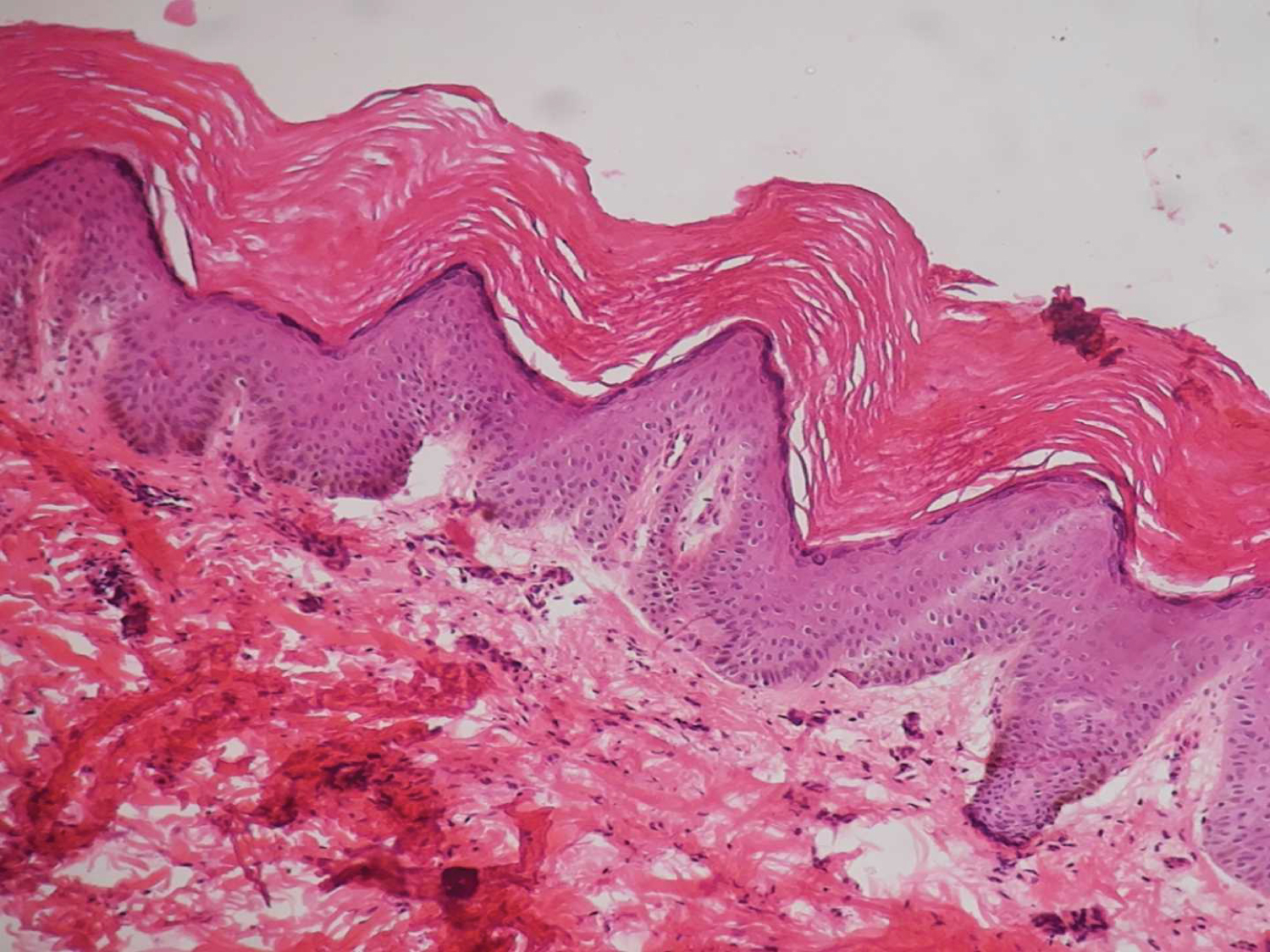 Figure 2: Sections examined show epidermis with hyperkeratosis, acanthosis and hypergranulosis, and papillomatosis with a "church spire" pattern (H&E, ×20).
View Figure 2
Figure 2: Sections examined show epidermis with hyperkeratosis, acanthosis and hypergranulosis, and papillomatosis with a "church spire" pattern (H&E, ×20).
View Figure 2
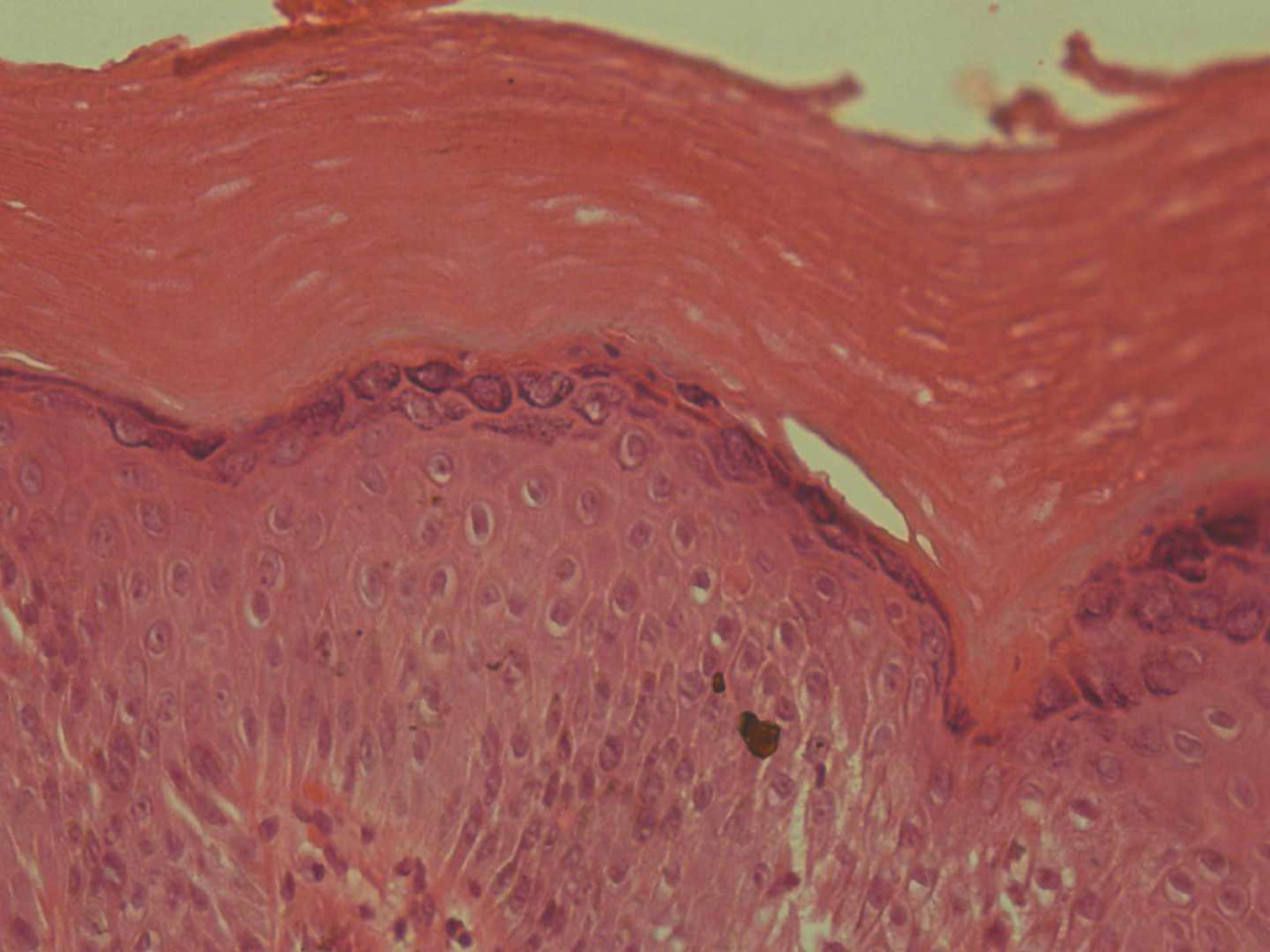 Figure 3: The section examined shows pure compact hyperkeratosis, acanthosis, and papillomatosis without parakeratosis, dyskeratosis, and vacuolization (H&E, ×40).
View Figure 3
Figure 3: The section examined shows pure compact hyperkeratosis, acanthosis, and papillomatosis without parakeratosis, dyskeratosis, and vacuolization (H&E, ×40).
View Figure 3
AKV is a rare genodermatosis that develops at birth or during early childhood. However, non-familial cases like our case have been reported in the literature [4,5]. Characteristic findings of AKV include asymptomatic flesh-coloured or reddish-brown flat papules resembling flat warts. Typically it arises in the dorsal aspect of the hands, and feet. It may extend to the forearms and lower legs. Punctate keratoses of the palms and soles have often been detected in AKV. Clinically, they can be confused with verruca plana, lichen planus, epidermodysplasia verruciformis, especially with Darier’s disease [1].
The pathogenesis behind AKV is the same as that of Darier's disease with minor allelic variation. The P602L mutation in ATP2A2 leads to defective calcium transport in the endoplasmic reticulum. This defect causes disordered keratinization leading to verrucous lesions and papules on the hand and feet. Given a dominant mutation, the disease has a chronic course without remission [1]. There have been reports of HPV-17 association with AKV and rarely malignant transformation to squamous cell carcinoma [5].
Histopathologically, AKV shows an exaggeration of the epidermal layer and thickening of the granular layer. These layers show a blunt domed pattern within concavities of the wavy stratum corneum, a so-called “church spire” [6]. There can be some degree of acantholysis, but not parakeratosis, dyskeratosis, or basal layer change. Clinically mimicking diseases can be ruled out with histopathologic findings. The hypertrophic variant of seborrheic keratosis might present an identical histological finding; however, clinical findings of seborrheic keratosis are far from those of AKV [7].
As a result of similar clinical and histologic findings, some authors have suggested a possible relationship between AKV and Darier’s disease. Histopathologically, AKV may be distinguishable from Darier’s disease. The unique finding of AKV is a “church spire”; otherwise, Darier’s disease is benign dyskeratosis with corps round [1].
In terms of the clinical features and histopathological findings, there is no difference between familial AKV and non-familial cases. Clinically, both show asymptomatic flesh-coloured to reddish-brown flat papules resembling flat warts on the dorsal palm and trunk. In addition, there is a histologically unique finding of a “church spire” [6]. In our case, the late onset age and absence of a familial history were confusing; however, with the typical clinical and histopathological findings, we were able to diagnose our patient as non-familial AKV.
We found eight previously reported cases of sporadic AKV in English literature (Table 1). Like ours all these cases showed typical clinical and histopathological findings and lacked a familial history. The onset age of classical AKV is different from that of sporadic AKV. Classical AKV often occurs during childhood; however, the onset age of sporadic AKV is much later than that of classical AKV. In addition, palmar and plantar keratoses have frequently been reported in classical AKV5, 12, but not in sporadic AKV.
Table 1: Sporadic cases of AKV. View Table 1
The differentials for AKV include EDV, Darier disease, verruca plana, lichen planus, and seborrheic keratosis. However, the lack of crops and roads, viral cytopathic changes in the granular layers, lichenoid changes, and comedo-like openings with milia cysts help diagnose AKV [1,7].
Superficial ablation has been reported to be effective in AKV. Other methods include cryotherapy, laser therapy, keratolytic solution, and surgical excision. The use of oral isotretinoin has been suggested. Regular follow-up is needed to prevent the possibility of a rare chance of transformation to squamous cell carcinoma [1,8,9].
AKV is a rare genodermatosis that usually develops in childhood but has rarely been reported in adulthood. Histopathologically, these lesions show considerable hyperkeratosis, acanthosis, and papillomatosis, mimicking a “church spire”, and a thickened granular layer. Superficial ablation has been reported to be effective.
Not applicable.
Written informed consent was obtained from the patient to publish this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
All the data regarding the findings are available within the manuscript.
The authors declare that they have no competing interests.
Not applicable.
TS carried out concepts & design, literature search, participated in clinical study and will stand as guarantor also. MKP carried out data acquisition, data analysis & manuscript preparation. All the authors have read & approved the final manuscript.
Not applicable.