Congenital pulmonary airway malformations (CPAMs) consist of a spectrum of cystic or non-cystic lung malformations often associated with bronchial atresia during in utero development. Several classifications for these lesions have been proposed using radiologic or pathologic criteria based on cyst size and histology, with the Stocker classification being perhaps the most commonly used in the pathology literature. However, those classifications of CPAM have relied predominantly on observations made in either postnatal lung resections or stillborn infants at autopsy and generally not applicable to prenatally resected CPAMs. We present a case of CPAM with intra-lobar sequestration in a 15 weeks old fetus. To the best of our knowledge, it is the first case reported in such an early developmental stage. There were some difficulties in classifying it due to limited pathologic classification schemes suitable for fetal lungs. More pathologic studies of pulmonary malformations occurring during the fetal period are required to highlight the variability of findings seen in such lesions at all developmental stages.
Congenital pulmonary airway malformations (CPAMs)consist of a spectrum of cystic or non-cystic lung malformations often associated with bronchial atresia during in utero development. Several classifications for these lesions have been proposed using radiologic orpathologic criteria based on cyst size and histology, with the Stocker classification being perhaps the most commonly used in the pathology literature [1,2]. CPAMs have been classified by Stocker according to cyst size and histological resemblance to segments of the developing bronchial tree and airspaces. The newer classification scheme includes five types and only three types of CPAMs are distinguished at imaging: large cyst CPAM (type I) and small cyst CPAM (type II), which constitute macrocystic CPAMs; and microcystic or solid type (type III) lesions, which have cysts that are smaller than 5 mm in diameter, with no discernible cystic spaces. Most CPAMs derive their blood supply from the pulmonary artery and drain via the pulmonary veins with the exception of some cases of macro cystic CPAMs, especially small cyst CPAMs (type II). Such lesions are often referred to hybrid lesions and demonstrate histological and imaging features of both a CPAM and broncho- pulmonary sequestration [3]. However, current descriptions and classifications of CPAM have relied predominantly on observations made in either postnatal lung resections or stillborn infants at autopsy, while fetal lung malformations have been described radio graphically may change character over time and even regress. This suggests a potential difference between those lesions observed in prenatal versus postnatal life and makes these classification schemes generally not applicable to intrauterine cystic lung lesions resected during fetal life [4].
A 30-year-old patient, gravida 2 para 1 presented at 15 weeks of gestation. The parents were generally healthy with no remarkable family history. Nuchal translucency scan was normal. Sonographic examination revealed an appropriate for gestational age male fetus. Mild pericardial effusion and ascites were evident and an inverted A wave was demonstrated in the ductus venosus consistent with fetal cardiac insufficiency. The left apical heart was shifted to the right and the stomach was in a central position (mesogastria). The left lung was comprised of an echogenic mass with a measured circumference in an axial plane of 91 mm. No macro-cystic lesions were evident (Figure 1a, Figure 1b). Two large vessels exhibiting systemic venous and arterial flow were demonstrated entering the mass to form a vascular plexus (Figure 1c, Figure 1d, Figure 1e, Figure 1f). The diaphragm was flattened by the enlarged left lung. The right lung was compressed backwards and small (measured circumference in an axial plane 48 mm). A detailed anatomical scan did not reveal any further malformations. Following a comprehensive consultation regarding the findings, the parents elected termination of pregnancy.
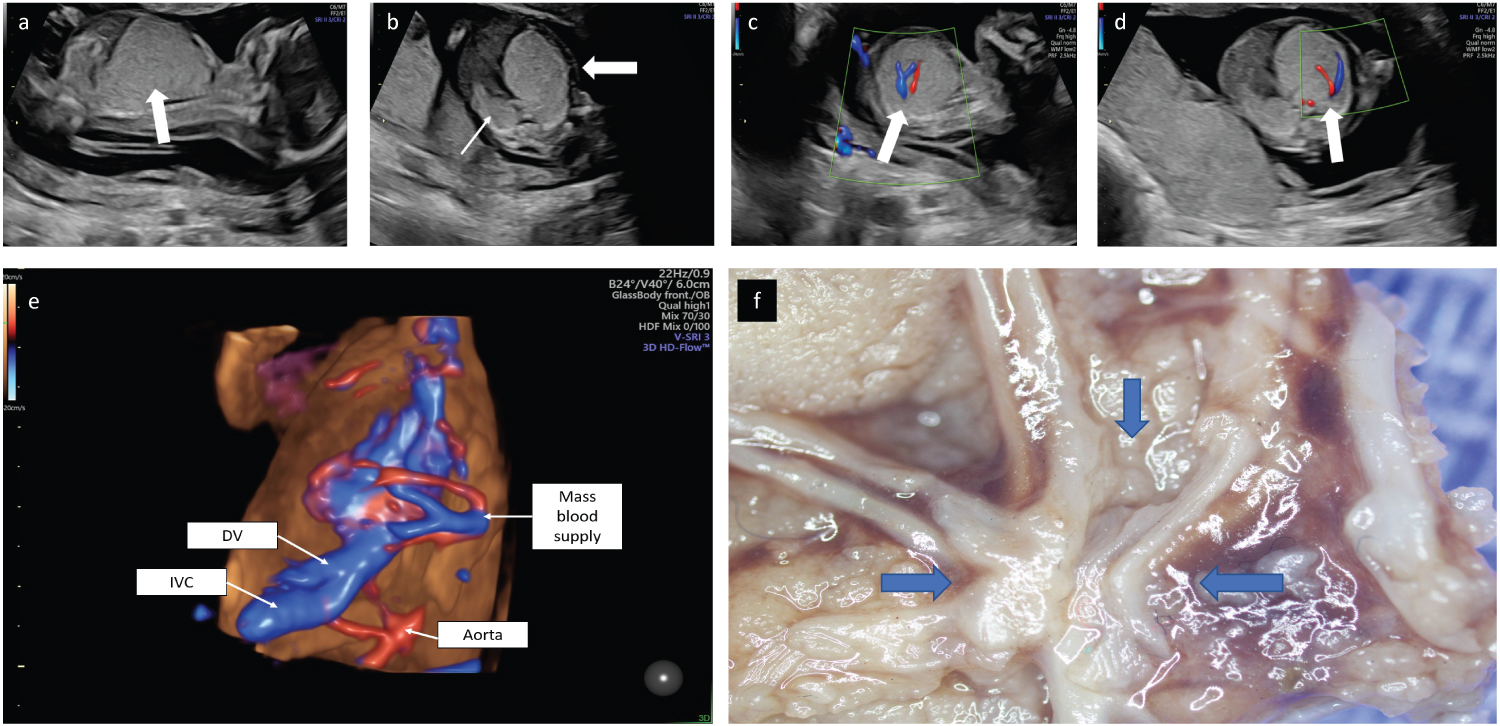 Figure 1: a) 2D ultrasound obtained at 15.4 weeks of gestation. Mid-sagittal plane demonstrating an echogenic mass that comprised the left lung with a caudal mass effect, compression of the diaphragm and abdominal organs. b) Axial view demonstrating the mass effect leading to a right shift of the heart (bold arrow). The right lung is small and compressed posteriorly (thin arrow). c) 2D Doppler flow in a parasagittal view demonstrating a large artery and a large vein at the center of the mass (arrow). d) 2D Doppler flow axial view demonstrating a large artery and vein at the center of the mass (arrow). e) 3D Doppler flow of the vessels within the thoracic mass. f) Stereotactic dissection demonstrated a plexus of aberrant vessels within the mass.
View Figure 1
Figure 1: a) 2D ultrasound obtained at 15.4 weeks of gestation. Mid-sagittal plane demonstrating an echogenic mass that comprised the left lung with a caudal mass effect, compression of the diaphragm and abdominal organs. b) Axial view demonstrating the mass effect leading to a right shift of the heart (bold arrow). The right lung is small and compressed posteriorly (thin arrow). c) 2D Doppler flow in a parasagittal view demonstrating a large artery and a large vein at the center of the mass (arrow). d) 2D Doppler flow axial view demonstrating a large artery and vein at the center of the mass (arrow). e) 3D Doppler flow of the vessels within the thoracic mass. f) Stereotactic dissection demonstrated a plexus of aberrant vessels within the mass.
View Figure 1
At autopsy aborted male fetus weighing 200 gm appeared appropriate for gestational age and showed mild signs of maceration and autolysis. Internal examination disclosed an immense mass in the left lower lobe (Figure 2a) resulted in midline shift to the right, hypoplasia of right lung, pericardial effusion (2 ml), ascites (4 ml) and no other abnormalities. The mass was measured 3 × 4 × 5 cm and composed of spongiform cystic grey tissue. Histological examination of the lesion revealed unevenly sized airspaces (Figure 2b) measured up to 6 mm in diameter lined by columnar epithelium with clear to pink cytoplasm, central to apical nuclei, and frequent sub-nuclear vacuolization (Figure 2c). Prominent smooth muscle bundles were underlying most of the airspaces, this was highlighted by immunostaining for smooth muscle actin (Figure 2d). The spaces were surrounded by abundant cellular mesenchymal tissue resembling immature lung parenchyma. Hilar connective tissue contained bronchi with cartilage and disproportionally small accompanying arteries. Mucus cells, skeletal muscle, and non-bronchial cartilage were not seen within this lesion.
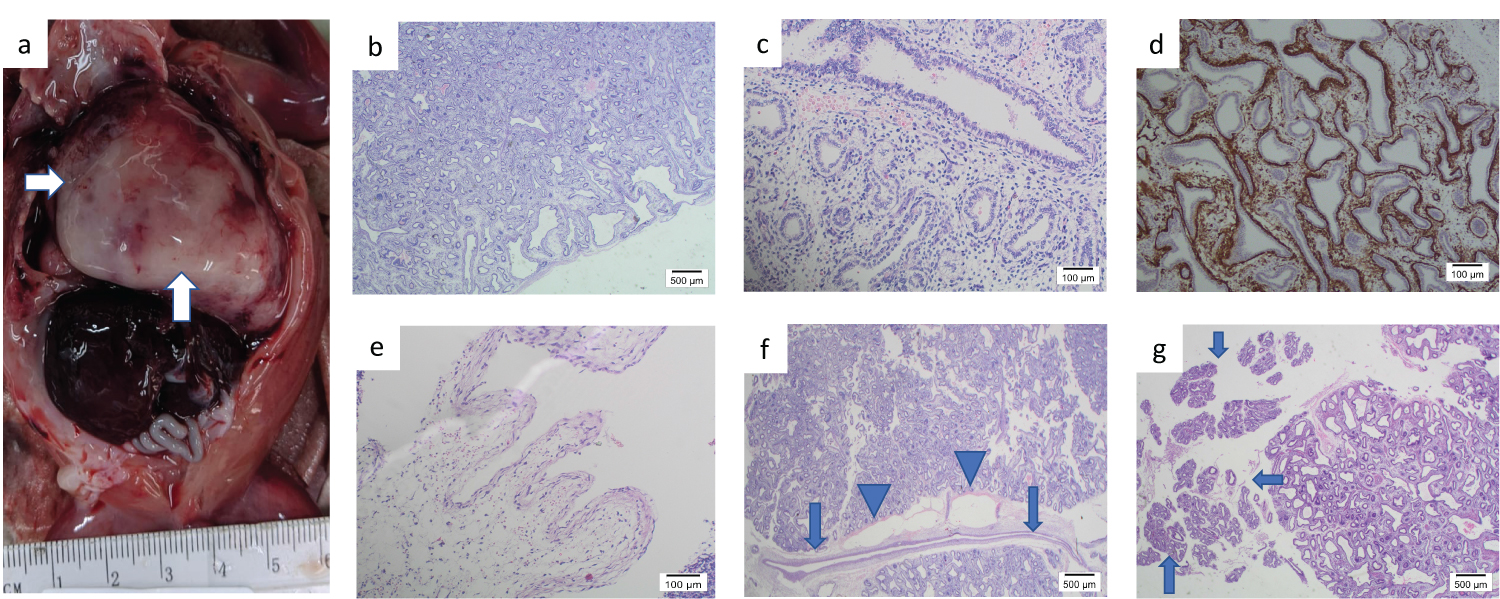 Figure 2: a) The mass in the left lower lobe (arrows). b) H & E x1. Unevenly sized airspaces measured up to 6 mm in diameter. c) H & E x10. The airspaces lined by columnar epithelium with clear cytoplasm, central to apical nuclei, and frequent subnuclear vacuolization. d) SMA x4 Smooth muscle bundles underlying most of the airspaces highlighted by immunostaining for smooth muscle actin. e) H&E x10. Dilated ornate venous channels. f) H & E x1. An aberrant artery without ramification for at least 10 mm (arrow) with markedly dilated lymphatics (arrowhead) in the hilar region. g) H & E x1 Normal lung tissue in the pseudo glandular stage of development in the periphery of the lesion (arrows).
View Figure 2
Figure 2: a) The mass in the left lower lobe (arrows). b) H & E x1. Unevenly sized airspaces measured up to 6 mm in diameter. c) H & E x10. The airspaces lined by columnar epithelium with clear cytoplasm, central to apical nuclei, and frequent subnuclear vacuolization. d) SMA x4 Smooth muscle bundles underlying most of the airspaces highlighted by immunostaining for smooth muscle actin. e) H&E x10. Dilated ornate venous channels. f) H & E x1. An aberrant artery without ramification for at least 10 mm (arrow) with markedly dilated lymphatics (arrowhead) in the hilar region. g) H & E x1 Normal lung tissue in the pseudo glandular stage of development in the periphery of the lesion (arrows).
View Figure 2
Stereotactic dissection discovered a plexus of vessels within the mass (Figure 1f) that led to the hilum of the lobe. Aberrant vessels including markedly dilated ornate venous channels (Figure 2e) and straight non-branching arteries within fibrous septa without accompanying bronchi were noted. One of those arteries without ramification for at least 10 mm was seen in the hilar region (Figure 2f). The aberrant vessels were accompanied by markedly dilated lymphatics (Figure 2f). No other vessels apart from hilar ones were identified. There was normal lung tissue in the pseudo-glandular stage of development in the periphery of the lesion (Figure 2g). There were no signs of bronchial obstruction such as airway mucostasis or foamy macrophages.
The majority of lung cystic lesions are diagnosed at 18-20 weeks gestational age during routine prenatal ultrasound imaging [5], and to the best of our knowledge there are no any published cases till 18 Weeks of gestational age (WGA).We faced difficulties in categorizing the lesion because it didn't meet the criteria of Stocker classification likely because it has relied predominantly on observations made in either postnatal lung resections or stillborn infants at autopsy [2]. Classification scheme suitable for fetal lung malformations were required. Kreiger and others offered a novel classification based on surgical resections made from 21 to 38 WGA including those resembling the pseudo-glandular period of fetal lung development [4]. The presented lesion consists of cystic unevenly sized airspaces lined by columnar epithelium with frequent sub-nuclear vacuolization and smooth muscle bundles underlying most of the airspaces. Mesenchymal tissue resembling immature lung parenchyma was noted between the cystic spaces. These findings are compatible with the features described in 1st and 2nd groups of the above-mentioned classification. It was lack of relatively mature-appearing alveolar-type air spaces lined bycuboidal to flattened epithelium with pink to clear non-vacuolated cytoplasm to define it as Group III. In addition, the lesion has aberrant vessels that seem to enter the lesion via the hilum and actually appears to be so called "hybrid lesion". However, an abnormal systemic arterial supply can be seen in some cases of CPAMs, especially small cyst CPAMs [3]. Thus, we classified the lesion as a mixture of 1st and 2nd Groups according to Kreiger and others classification in the absence of any other option.
With advances of early detection of pulmonary malformations by in utero radiographic imagining that leads either to resection of such lesions during fetal life or termination of pregnancy the newer pathologic classification schemes applicable to prenatally resected CPAMs are needed. Therefore, more work should be carried out to further characterize the histopathology of prenatally resected CPAMs in early stages of development.