Acute necrotizing encephalopathy (ANE) is a rare clinical-imaging syndrome with unknown etiology, characterized by acute fulminant severe encephalopathy and brain damage with multifocal symmetry. ANE has no specific clinical symptoms and signs, similar to common encephalitis or encephalopathy symptoms. The characteristic brain imaging examination is diagnostically significant. To date, no specific treatment for ANE is available and the prognosis is poor. Here, we reported a typical case of ANE in a child, in whom good outcome was achieved via combined therapy with immunoglobulin and glucocorticoids.
Acute necrotizing encephalopathy, Children, Case, Outcome
An eight-month, two-day-old male child had fever, diarrhea and vomiting three days before admission. He then become lethargic and irritating after arousal with repeated tonic-clonic movements on the day prior to admission. For further management, he was transferred to our hospital with continuous intravenous infusion of midazolam. The child had normal intellectual and motor development prior to the illness onset. His parents denied family history of dyskinesia or mental retardation. The physical examination on admission showed no abnormalities except that he had increased muscle tone in his limbs and right hemiplegia after stimulation. His bilateral Babinski's signs were positive. Serum biochemistry revealed white blood cell count 1.91 × 109/L, neutrophils count 0.59 × 109/L, calcitonin 25.41 ng/ml, serum glutathione transaminase 71.8 U/L. Renal function, electrolytes, coagulation, blood glucose, blood ammonia, lactic acid and C reactive protein were normal. Serum IgM antibody against Coxsackie virus B was positive, while the antibodies against Epstein-Barr Virus, herpes simplex Virus and enterovirus 71 were negative. Antibodies against Respiratory Syncytial Virus, adenovirus, influenza virus and parainfluenza virus were also negative. Bacterial culture of blood and sputum were negative. The concentration of microalbumin in the cerebrospinal fluid (CSF) was 0.52 g/L. No other abnormalities were found in routine CSF parameters, viral antibodies or bacterial culture of CSF; neither were there abnormalities on blood tandem mass spectrometry or organic acids in urine. On EEG, background δ and θ activity (2-4 Hz) was found during wakefulness. Brain Magnetic Resonance Imaging (MRI) on the 4th day of onset revealed a wide symmetrical abnormal signal in the brain, primarily in the thalamus (Figure 1a). No mutation highly-related to Acute necrotizing encephalopathy (ANE) was found on Trio clinical exon sequencing or mitochondrial DNA detection.
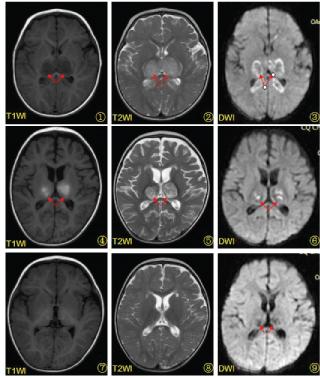 Figure 1: a) MR imaging on the fourth day of the patient's course with symmetrical abnormal signals in the bilateral thalamus; b) MR imaging on the eleventh day of the patient's course with more localized lesions and hemorrhagic signals in the bilateral thalamus; c) The results of reexamination 2 months after onset revealing that the areas of lesions are significantly reduced. T1WI T1 weighted imaging, T2WI T2 weighted imaging, DWI diffusion-weighted imaging.
View Figure 1
Figure 1: a) MR imaging on the fourth day of the patient's course with symmetrical abnormal signals in the bilateral thalamus; b) MR imaging on the eleventh day of the patient's course with more localized lesions and hemorrhagic signals in the bilateral thalamus; c) The results of reexamination 2 months after onset revealing that the areas of lesions are significantly reduced. T1WI T1 weighted imaging, T2WI T2 weighted imaging, DWI diffusion-weighted imaging.
View Figure 1
The child was initially treated with a combination of vancomycin, meropenem, mannitol, glycerol and fructose in the local hospital on the day prior to transfer. After admission, the child was treated with combined therapy including gamma globulin(IVIG)1 g/kg daily for two days, methylprednisolone 10 mg/kg daily for five days followed by oral prednisone acetate 1.5 mg/kg daily. Mannitol and fructose glycerol were given to reduce intracranial pressure with the adjunctive treatment of coenzyme Q10, L-carnitine and vitamin B. We did not use any other antiepileptic drug because his seizure was controlled just after admission. We used some diphenhydramine and clonazepam to relieve his muscle tone during the recovery period.
The state of consciousness was improved just after IVIG and 5 days of methylprednisolone. MRI re-examination on the 11th day after onset showed the wide range of brain abnormalities became more focal than prior, accompanied by hemorrhagic foci in bilateral thalamus and corpus callosum (Figure 1b). The signs of provocation, muscle tone and hemiplegia of the right limb all improved gradually, and he was discharged 22 days after onset. The follow-up after two months suggested a four-month lag behind his peers in physical ability with tremor of the right upper limb. He had mild hemiplegia with no laterality of the deep tendon reflex or Babinski reflexes. Nevertheless, his cognitive level was near normal. A repeat electroencephalogram found no epileptic discharges. A repeat MRI also revealed that the lesions had significantly absorbed (Figure 1c). We had the 10-month follow-up and found that he had no difference in motor and language cognition skills than his peers except for mild paralysis of the right upper limb with grade Ⅳ muscle strength.
Acute necrotizing encephalopathy (ANE) is a rare clinical imaging syndrome with unknown etiology characterized by acute fulminant severe encephalopathy and multifocal symmetrical brain damage. Cases have been reported all over the world, with more cases in Asia since the first case was discovered by the Japanese scholar Mizuguchi in 1995 [1]. ANE is primarily secondary to viral infection and occurs throughout the year, especially during flu season. The peak age in infants is six to eighteen months, but it is also observed in adolescents and adults. Associated pathogens may include influenza A and B, new influenza A virus (H1N1), parainfluenza virus, herpes simplex virus, varicella-zoster virus, human herpesvirus 6 (HHV-6), coxsackie virus, rotavirus, measles virus, rubella virus, mycoplasma pneumoniae and others, however, influenza virus is the most common [2-4]. Some studies reported no differences in clinical course or prognosis of ANE between those subtypes caused by influenza virus and those caused by non-influenza virus [5]. Therefore, it is thought that the progression of ANE is not dependent on the type of viral infection.
ANE is not caused by direct infection of virus, and its pathological changes primarily include damage to the blood-brain barrier caused by focal vascular injury, plasma exudation, brain edema, dot hemorrhage and necrosis of neuronal and glial cells. Gross pathological findings show symmetrical encephalomalacia accompanied by partial dissolution of brain, primarily found in the thalamus, brainstem tegmentum and deep white matter of the brain and cerebellum [6]. The pathogenesis of ANE remains unknown; the hypercytokinemia hypothesis, i.e. the hyperimmune response induced by cytokine storms, is the most common hypothesis regarding pathogenesis [7]. Cytokine storms cause brain damage by altering vascular permeability, leading to multiple-organ damage, e.g. liver damage, acute kidney damage or diffuse intravascular coagulation. In some cases, hemophagocytic syndrome is part of the clinical presentation. In addition, metabolic disorders or mitochondrial dysfunction may be involved in the pathogenesis, explaining why brain injury lesions primarily involve anatomical sites with high energy metabolism, including the thalamus and brain stem.
ANE occurs sporadically in most cases. A higher proportion has been reported in Asia, especially in Japan. This may be ascribed to the genotypes of human leukocyte antigen (HLA) in the region [8]. Mutations of RNABP2, SCN1A and CPT2 genes may be associated with the pathogenesis of ANE [9-11]. Familial or recurrent ANE cases (named ANE1) have been reported in recent years, and the gene mutation of RNABP2 was demonstrated to play an important role in ANE1 cases [3,12]. As the exogenicity rate of RNABP2 gene is only 40% [12], the occurrence of ANE should be the result of interactions of genetic predisposition and environmental factors. In the present case, the child had no family history of ANE. His illness was likely triggered by a gastrointestinal infection based on positive IgM antibody against Coxsackie B virus. We hypothesized that the morbidity would be linked to the viral infection against a certain genetic background. Nevertheless, no mutation associated with ANE was identified on exon sequencing or mitochondrial DNA detection, thus the case should be regarded as sporadic.
ANE has no specific clinical symptoms or signs, and in this regard, it is similar to common encephalitis or encephalopathy. Some scholars classified evolution of ANE into prodromal stage, acute encephalopathy stage and convalescent/chronic stage [1,13]. As many as 90% of the children may have precursor symptoms of viral infection, including upper respiratory tract infection, viral gastroenteritis, infantile rash or others. The symptoms of acute encephalopathy occur one to three days after the prodromal infection. As many as 94% of the cases have early seizure activity most frequently persistent tonic-clonic seizures. Almost all (98%) of the patients develop rapid consciousness impairment and possible progressive exacerbation, with Glasgow scoring ranging from 6 to 7. It can be accompanied by frequent vomiting (70%), papillary edema (38%), early onset of cerebrospinal ankylosis (85%), pupil narrowing (73%), positive Babinski's sign (66%) and tendon hyperreflexia (66%). The condition may deteriorate rapidly during this period, manifesting as signs of sustained high fever ranging from two to five days, decreased muscle tension, frequent apnea, dilated pupil, hypotension and even some fatal complications of diffuse intravascular coagulation, multiple organ dysfunction and hemophagocytic syndrome. About 30% of patients died during this period. Some patients may also have spinal cord involvement, with signs of transverse myelitis [14,15]. The convalescence period, marked by the restoration of consciousness, is achieved after the onset of six to ten days if the condition does not deteriorate. The recovery of nervous system function requires several months, and fewer than 10% of patients achieve complete recovery. Most patients are left with various degrees of sequelae, from lesser conditions of mild hemiplegia, intentional tremor, athetosis, abductor nerve palsy and strabismus induced by abnormal extraocular muscular activity, to more serious signs, including mental retardation, epilepsy, quadriplegia and persistent vegetative state. The characteristics of our patient's clinical course was consistent with progression of ANE. Compared to previously described cases, his fever receded faster and the body temperature returned to normal when encephalopathy developed even if we didn't use some special treatment related to temperature restoration. The severity of his consciousness disorder was also not severe, only manifesting as lethargy. His seizure was easily controlled without antiepileptic drugs. His recovery was relatively good. We speculate that It should be borne in mind that the degree of fever, consciousness disorder and difficulty of seizure controls may be related to the prognosis in addition to therapeutic factors.
Blood tests in ANE show elevated white blood cell counts with a dominant proportion of neutrophils, and concentrations of CRP and ESR are also elevated. Severely affected patients may suffer from DIC, manifesting as a combination of thrombocytopenia, prolonged prothrombin time, decrease of fibrinogen and increase of fibrinolytic degradation products. The concentrations of serum transaminase, LDH, creatine kinase (CK) and urea nitrogen also increase. Hypoproteinemia may develop in some patients, while high blood ammonia, hypoglycemia and electrolyte disturbances rarely occur [16]. Tests of cerebrospinal fluid show no specific alterations except for the increase of protein concentration. The electroencephalogram in the acute phase shows widespread slow waves, and its background activity could show a gradual improvement with recovery of consciousness.
Brain imaging is diagnostically significant [17]. It is characterized by symmetrical multifocal lesions primarily localized in the thalamus (100%), superior brainstem tegmentum (61%), periventricular white matter (56%) and cerebellar medulla (51%), while the anterior part of the lentiform nucleus, periaqueductal gray matter, optic nerve, substantia nigra and inferior olivary nucleus are rarely involved. Computed tomography (CT) images show symmetrical decreases in density at the lesion sites. MRI reveals early impairment of gray matter with low signal on T1 weighted images and high signal on T2 weighted images. The T1 weighted images of thalamus show signs of concentric rings three days after the onset of encephalopathy, which is high signal in the central lesion but low signal in the periphery. Subacute hemorrhage appearing as high signal rings on T1 image can be detected in thalamic lesions in the second week after onset. Impairments of white matter are always low signals on T1 but high signals on T2 weighted images. The apparent diffusion coefficient (ADC) of diffusion-weighted imaging better reflects the pathological changes, the typical manifestation of which is the characteristic "trichrome pattern", i.e., a higher ADC value in the central region of the thalamus (hemorrhagic necrosis) with a surrounding lower ADC value (cytotoxic brain edema) compared to normal brain tissue. The periphery of the lesion has a higher ADC value than does the center which is angiogenic brain edema. The center of the lesion in mild cases is free of hemorrhage or necrosis. Its ADC image shows a relatively low value in the central part (cytotoxic brain edema) but a relatively high value in surrounding area (angiogenic brain edema). The lesions shrink in the chronic and convalescent stages. Severe cases may develop multiple cysts with hemosiderin deposits and brain atrophy. Several reports mention about outcome with ANE related to with/without brainstem lesions. This case did not show brain stem lesions, it might be somewhat related his well outcome.
No specific treatment for ANE is currently available. The conventional treatment is primarily supportive with anti-cytokine therapy, as ANE is thought to be cytokine-mediated. The therapeutic objectives in the acute phase are seizure control, lowering intracranial pressure via inhibiting brain edema and maintaining good ventilation and cerebral perfusion. Hypothermia therapy [18] and immunotherapy with methylprednisolone (30 mg/kg/d), gamma globulin or plasma exchange [19] may be effective. The combination of gamma globulin (400 mg/kg) given once a month with long-term oral dexamethasone (0.075 mg/kg/d) was shown effective to prevent recurrences in patients with ANE1 [20]. To date, no consensus has been reached regarding timing and length of glucocorticoid treatment. In our case, methylprednisolone was administered at 10 mg/kg/d in the acute phase, followed by oral prednisone at 1.5 mg/kg with gradual taper. The length of oral prednisone therapy lasted about three months and a beneficial therapeutic effect was achieved. No long-term oral glucocorticoid therapy was given, however, long-term follow-up is warranted because of the risk of relapse.
The prognosis of ANE is poor, with mortality rates as high as 30%. Most survivors have various degrees of sequelae. Fewer than 10% of the patients recover completely and the recovery of cognitive function is generally better than that of physical function. The prognosis may be worse if the patients have significantly elevated concentrations of serum aminotransferase and cerebrospinal protein. Patients with hemorrhagic lesions or brainstem involvement on image also have worse prognoses. The quantity of brain lesions negatively correlate with prognosis. Some researchers have established an ANE severity score (ANE-SS) [21] as follws: Three for stroke, two for brainstem lesions, two for age over 48 months, one for platelet count less than 100 × 109/L, and one for cerebrospinal protein over 60 mg/dl. The prognosis of ANE-SS 0-1 is good, while that of ANE-SS 5-9 is poor. The ANE-SS of our case was zero, suggesting good prognosis.
ANE is a rare syndrome with unknown etiology, no specific clinical presentation or treatment. Brain imaging characterized by symmetrical multifocal lesions is diagnostically significant. Good outcome can be achieved via combined therapy with immunoglobulin and glucocorticoids.
Here is no conflict of interest.