Primary ciliary dyskinesia, Children, Registry, Diagnosis, Management
Primary Ciliary Dyskinesia (PCD) is a rare, genetically heterogeneous and multi-organ disorder, caused by impaired structure and/or function of motile cilia. Impaired ciliary function results in recurrent upper and lower respiratory tract infections (due to ineffective clearance of mucous secretions and inhaled particles). Clinical manifestations of PCD are diverse and include recurrent and chronic infections in the lung, ear, nasal and sinus and involvement of other systems such as cardiovascular (congenital heart diseases and laterality defects such as situs inversus totalis) and reproductive systems (infertility). PCD is a rare inherited disease affecting 1:10,000-40,000 individuals [1]. In addition, PCD is difficult to diagnose. There is no single standard diagnostic test but rather a combination of tests, some of which require high expertise [2]. Due to the rarity of the disease there is currently insufficient information about its clinical characteristics, and thus PCD in children is under diagnosed and diagnosed too late particularly in relatively small centers with minimal or no experience with PCD patients [3,4]. This may lead to increased morbidity and mortality. While there is wealth of publications describing clinical presentation of PCD, awareness of PCD and sufficient diagnostic facilities in various non-central locations, may limit the quality of PCD diagnosis and management.
Patients registry is a well-known tool to assemble sufficient numbers of individuals with a rare disease and to assess and monitor their data in a standardized and longitudinal way, as well as to recruit candidates for clinical research studies. While PCD data bases have been established in Europe and in the US [3,5], there is no dedicated Canadian PCD registry. In order to better characterize the demographic characteristics, clinical symptoms, various diagnostic tools and management in our region, we decided to establish a provincial PCD registry in Alberta. This PCD registry may be replicated in more Canadian provinces to allow a better broad networking of centers treating PCD patients, create standardization of diagnostic algorithms and evaluate effectiveness of treatment throughout the provinces.
The Alberta PCD registry is a retrospective and prospective cohort study. The registry was initiated at the PCD clinic at the Stollery Hospital in Edmonton.
We used charts' review and patient's interviews. We collected information about demographic, clinical characteristics, diagnostic tests of PCD [such as Transmission Electron Microscopy (TEM), High Speed Video-Microscopy Analysis (HVMA), nasal nitric oxide (nNO) and genetic analysis], progression of the disease, infections pathogens colonization and management. All data were collected on Research Electronic Data Capture (REDCap) database. REDCap uses 128-bit SSL encryption between the user's browser and the server and complies with University of Alberta encryption policy by storing data on Faculty of Medicine servers. System access is controlled using REDCap's standard user access controls as configured and reviewed by the University's risk management and privacy groups. REDCap supports all necessary security requirements.
Inclusion criteria included patients of any age with PCD in Alberta. PCD was defined as recently suggested by the ATS guidelines [6]. Exclusion criteria included missing the ability to get written informed consent. The study was approved by University of Alberta Ethics board and is registered in a public clinical trial registry (NCT03271840) as well as in the RoPR (The Registry of Patient Registries). All patients or parents signed an informed consent form.
1. Administrative data- demographic characteristics: birth month, birth year, gender, ethnic origin, socio-economic background. 2. Family history: consanguinity, affected siblings, other affected family members. 3. Symptoms leading to diagnosis: history of neonatal respiratory distress syndrome, situs inversus, chronic rhinitis/rhinosinusitis, chronic/recurrent otitis media, chronic wet cough, chronic/recurrent pneumonia. In addition, details on congenital heart defect, cystic kidney disease, hearing loss, male infertility, hydrocephalus, retinitis pigmentosa were also sought. 4. Age at diagnosis and results of the following diagnostic tests as performed: HVMA, TEM, nNO measurement and genetic analysis (for detailed description of the tests methods see Online Supplemental Material) 5. Vitality status.
1. Date and growth measures including height and weight 2. Updated clinical manifestations from all affected organ systems: upper airways (otitis media, hearing impairment, sinusitis, chronic rhinitis, nasal polyps), lung disease (haemoptysis, chronic cough, infectious exacerbations, pneumothorax) and comorbidities (OSA, GERD). 3. Microbiology studies: sputum cultures, throat swab, nasal swab, ear swab and BAL. 4. Imaging results: CXR/CT- bronchiectasis/atelectasis/infiltrates and location. 5. Lung function assessment- spirometry results. 6. Management and therapeutic interventions including: antibiotics (systemic/inhaled), inhalation therapy (NaCl 0.9%/hypertonic saline/bronchodilator or inhaled corticosteroids), other therapies such as Singulair or macrolides, airways clearance therapy, oxygen therapy, long term mechanical ventilation, lung surgery (lobectomy/lung transplantation), upper airway surgery (tympanostomy/myringotomy tube insertion, adenoidectomy, sinus surgery, mastoidectomy). 7. Fertility in male/female (> 18y).
The data was analysed by using descriptive statistics. All parameters were described as arithmetic mean, standard deviation, median, minimum, maximum, and frequency distribution. As no null hypothesis was established for this study, no test statistical tests were employed.
The study was initiated in late 2017 and we recruited to date twelve patients with PCD. All patients presented to the PCD clinic at the SCH in Edmonton.
Of 12 patients presented to the clinic, 9 patients resided in the Edmonton area (central and northern Alberta) and 3 in Calgary area (southern Alberta). We demonstrate a slightly female predominance. The ethnic origin was Caucasian for 7 patients and middle eastern origin for 5 patients. Most of the patients had positive family history of PCD and 3 patients had a history of parenteral consanguinity. The age at diagnosis range from 1-18 y with a mean of 4.7 y. Demographic characteristics of the patients are summarized in Table 1.
Table 1: Demographic characteristics. View Table 1
The most common symptoms that led to diagnosis included chronic wet cough, which was reported in all patients. Chronic/recurrent otitis media was reported in 11 patients (92%) as well as chronic rhinitis/rhinosinusitis. History of neonatal respiratory distress syndrome was reported in 9 patients (75%), followed by hearing loss in 8 patients (66%). Less frequently chronic/recurrent lower respiratory tract infections (LRTI) were reported in 4 patients (33%), laterality defects were reported in 3 patient (25%) and chronic atelectasis was reported in 1 patient (8%). The percentage of clinical symptoms that led to diagnosis are shown in Figure1.
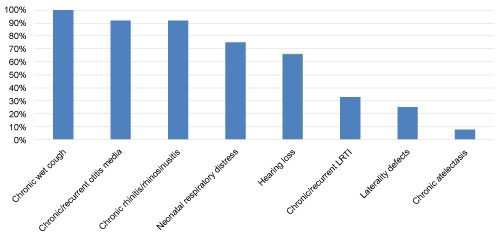 Figure 1: Percentage of clinical symptoms that led to diagnosis. View Figure 1
Figure 1: Percentage of clinical symptoms that led to diagnosis. View Figure 1
On physical examination, we demonstrated crackles (coarse), clubbing and wheezing in 9 patients (92%), 6 patients (50%) and 1 patient (8%) respectively.
Bronchiectasis on CXR or chest CT was seen in 6 patients (50%) whereas atelectasis was seen in 7 patients (58%) and Infiltrate in 8 patients (66%). Some of the patients had more than one finding.
Sputum cultures and BAL cultures were positive in 6 patients (50%) with Haemophilus influenzae as the most common pathogen. Other pathogens were Pseudomonas aeruginosa, Strep pneumonia, Neisseria meningitides, Staph aureus, Branhamella, Moraxella, Aspergillus fumigatus, Yeast and Cladosporium.
The microbiology culture results are shown in Table 2.
Table 2: Microbiology cultures results. View Table 2
Our institute has a diagnostic center for PCD that performs all of the currently recommended diagnostic tests for PCD including: nNO measurements using a Stationary Chemiluminescence machine (CLD 88 SP, Eco Physics Duernten, Switzerland), HVMA-which evaluates the function of the cilia, TEM to evaluate the ciliary ultrastructure and Genetic Testing.
Eleven patients had nNO measurement. nNO measurements were recorded in parts per billion (ppb) and converted to nNO production rate in nL/min with the following equation: nNO (nL/min) = NO (ppb) × 1/1000 × sampling flow (= 330 mL/min with our machine). All patients had abnormal results (lower than 77 nL/min). The lowest result was 1.5 nL/min and the highest result was 33 nL/min. The mean result was 13.8 nL/min. nNO measurement results are shown in Figure 2.
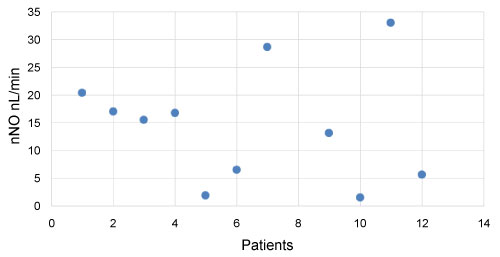 Figure 2: Nasal NO measurement results (nL/min). View Figure 2
Figure 2: Nasal NO measurement results (nL/min). View Figure 2
Five patients (out of 12) had HVMA testing. Two of them (40%) had insufficient samples and three of them (60%) showed abnormal increased ciliary beat motion with conclusive abnormal beat pattern.
Ten patients (out of 12) had TEM testing. Two of them (20%) had outer dynein arm and inner dynein arm (IDA) defects, one patient (10%) had tubular disorganization defect and one patient (10%) had previous reported test that was interpreted as "compatible with immotile cilia syndrome" (with no other details). Two patients (20%) had inconclusive results, two patients (20%) showed normal results, and two patients (20%) had insufficient samples.
All patients in the registry had genetic testing for PCD mutations, and all demonstrated pathological mutations, compatible with PCD. Five patients (42%) had CCDC39/40 mutation that causes an inner dynein arm (IDA) defect and axonal disorganization. Three patients (25%) had DNAI1 mutation, that causes- outer dynein arm (ODA) defect. Two patients (16%) had DNAH11 mutation with normal ultrastructure. Two patients (16%) had DNAAF2 mutation, that causes outer and inner dynein defects.
The diagnostic tests results including HVMA, TEM, nasal NO and genetic tests are shown in Table 3.
Table 3: Diagnostic tests results. View Table 3
Spirometry was performed in nine patients, 7 of them (78%) demonstrated obstructive pattern, mostly mild to moderate obstruction (86%). One patient (14%) demonstrated severe obstruction.
The spirometry results (all in percent predicted) are shown in Table 4.
Table 4: Spirometry results. View Table 4
Comorbidities were demonstrated in eight patients (67%), mostly gastroesophageal reflux and obstruction sleep apnea. ADHD, anxiety, allergy, asthma, chronic abdominal pain and hypertension were also present.
Management was supportive and nonspecific. The most commonly used treatments were bronchodilators, followed by inhaled corticosteroids, nasal steroids, systemic antibiotics (mostly amoxicillin/clavulanate and azithromycin) and chest physiotherapy. Hypertonic saline was used in one patient. Surgical treatment including lung surgery and ENT surgeries were also demonstrated. Medical and surgical management options are summarized in Table 5.
Table 5: Management options. View Table 5
We report here our first year of experience in developing the first provincial PCD registry in Canada. PCD is a rare genetically heterogenous disorder, characterized by motile cilia dysfunction causing chronic upper and lower respiratory tract infections, fertility problems and disorders of organ laterality [5,6]. PCD is mostly transmitted by autosomal recessive patterns of inheritance, however, rare cases of autosomal dominant and X linked inheritance patterns have also been reported [7]. There is high prevalence of PCD in societies with high degree of consanguinity [1] as was demonstrated in our study. To date there are 43 PCD causing known genes with many mutations that lead to several structural and functional cilia defects [1,6], five mutations were demonstrated in our registry. The median age at diagnosis in our cohort was 3.5y, slightly earlier than that reported in the European survey (5.3y).
Motile ciliated epithelial cells are present in the nasal cavity, paranasal sinuses, middle ear, airways, fallopian tube, cervix, vas deferens and ependyma. Ineffective ciliary function leads to abnormal mucociliary clearance and chronic bacterial infections. In addition, dysfunction of the cilia in the embryonic nose leads to laterality defects, whereas dysfunction of the cilia in the sperm flagella and fallopian tubes leads to infertility [8]. Literature suggests that clinical presentation of PCD in children include chronic productive cough (universal feature), recurrent pneumonia (≥ 60% of patients) chronic rhinitis/sinusitis (≥ 60% of patients) chronic otitis media (≥ 60% of patients) and conductive hearing loss (in 20-60% of patients) [7]. In addition, the majority of PCD patients have a history of neonatal respiratory distress (approximately 80% of patients) [6]. Situs inversus totalis is common in up to 55% of patients and heterotaxy in up to 12% of patients [6]. Bronchiectasis is reported in 20-60% of patients and clubbing in 20-60% [7]. Similar to the literature, the most common clinical symptoms in our patients were chronic wet cough, chronic rhinitis and recurrent otitis media. Many of them had a history of neonatal respiratory distress syndrome. However, the frequency of chronic/recurrent pneumonia was low, 4 patients (33%) for undetermined causes.
According to ATS guidelines for PCD diagnosis, there are 4 major clinical criteria for PCD phenotype [6]: 1. Unexplained neonatal respiratory distress in term infants. 2. Year-round daily cough beginning before 6 m of age. 3. Year-round daily nasal congestion beginning before 6 m of age. 4. Organ laterality defects. At least 2 of 4 criteria should be positive for PCD clinical phenotype and in their presence, the patient should be referred for further testing [6]. Without at least 2 of these clinical features, patients are unlikely to have PCD. All our patients met criteria for compatible clinical features with ten of them having 3 major clinical criteria, one having 4 major criteria and one having 2 major criteria.
Even in those subjects with compatible clinical features, definitive diagnosis can still be challenging and requires a combined approach of diagnostic tests, all of which have many limitations as describes below [5]. Measurement of nasal nitric oxide (nNO) is a very good screening test for PCD [5]. nNO production rate of less than 77 nL/min has a sensitivity and specificity of 0.98 and > 0.999, respectively, for PCD diagnosis [1,6]. The reason behind low nNO in PCD is unknown [8]. nNO should not be used as a single diagnostic test due to lack of specificity. The low levels can also be present in CF, sinusitis, nasal polyposis acute upper airways infections, HIV infection and smoking [5,6,8]. In addition, failure to obtain reliable nNO values can be secondary to nasal obstruction, equipment malfunction, high ambient nitric oxide or lack of patient cooperation [6]. Similar to the literature, all of our patients had abnormal results (lower than 77 nL/min).
Analysis of ciliary cross sections by TEM was once considered the gold standard for the diagnosis of PCD. However, TEM tests have many limitations and should not be used in isolation to exclude a PCD diagnosis. Nonspecific ciliary changes can be induced by exposure to environmental pollutants or infections and mimic PCD. In addition, 15-30% of affected individuals have normal ciliary ultrastructure on TEM. Furthermore, false positive diagnosis is common in subjects whose TEM showed isolated inner dynein arm defects [5]. Lack of experience in obtaining biopsy samples can lead to insufficient sample for TEM analysis, and lack of experience in processing and interpretation can lead to false positive or false negative results [6]. Similar to the literature, there were nonspecific results for TEM in our study.
High speed video microscopy of ciliary beat pattern (CBP) and ciliary beat motion of respiratory epithelial cells obtained by nasal brushing is commonly used in Europe for the diagnosis of PCD [5]. However, there is lack of standardization for HVMA sample preparation and interpretation among different centers [6]. In addition, nonspecific ciliary changes can be induced by infection and inflammation (secondary PCD) and mimic PCD. Furthermore, subtle beating abnormalities can be interpreted as normal [5]. Similar to the literature, there were nonspecific results for HVMA in our study.
As previously mentioned, 43 PCD genes with few hundred mutations that lead to several structural and functional cilia defects were identified so far [6]. Despite the dramatic increase in our molecular understanding of PCD, genetic tests are still expensive and commercially available panels include only 32 genes [6]. In addition, not all PCD mutations are known and genetic testing can detect only 65% of the cases [1,8]. Every year new genetic mutations are discovered, so negative results can't rule out PCD diagnosis [5]. The pathologic mutations found in our patients were in line with their microtubular ultrastructural defects and the cilia beat pattern.
Imaging studies in PCD patients are nonspecific and usually demonstrated consolidations, atelectasis and bronchiectasis. Bronchiectasis are more common in the middle lobe, lingula and lower lobes [5,7], a pattern observed also in some of our patients. Microbiological cultures of sputum or BAL usually present organisms that may be inhaled or aspirated from upper respiratory tract such as Haemophilus influenzae (65%), Staphylococcus aureus, Moraxella catarrhalis, Streptococcus Viridans and Streptococcus pneumoniae. Chronic Pseudomonas aeruginosa infection is more common in adults with advanced disease, as well as nontuberculous mycobacteria [1,5,7]. Similar to the literature, six of our patients had positive sputum or BAL cultures with Haemophilus influenzae as the most common pathogen. Pulmonary function tests may be normal during early childhood; however, they more commonly demonstrate obstructive airway disease (≥ 60% of the patients) [7]. Several new studies emphasize that lung function in PCD is affected from early age and shows a relationship between worse lung clearance index and microtubular ultrastructural defects. These studies emphasize the need to diagnose and treat the disease as early as possible, to try to minimize the progressive lung function decline [9,10]. Similar to the literature, seven of our patients presented with obstructive air flow in their spirometry.
Due to the rarity of the disease and the lack of clinical trials, management remains challenging and mainly supportive. The aim of PCD management is to maintain or recover lung function by early detection and aggressive management of complications [1]. The literature emphasizes the importance of enhancing airway clearance by chest physiotherapy and exercise [1].Aggressive antibiotic treatment is recommended in case of infection as well as management to prevent respiratory tract infections and irritation by routine immunization and avoidance of exposure to tobacco smoke or other irritants [5]. The literature also emphasizes the importance of regular surveillance including lung function testing, microbiological studies and review of airway clearing techniques is also suggested as well as CT/MRI to assess disease severity. Monitoring upper airway disease including regular hearing tests and assessment of sleep disordered breathing is also recommended [5,7]. Similar to the literature, management in our registry was supportive and nonspecific.
While we report here on the first PCD provincial registry in Canada, it is a single center study who collected data on PCD patients that arrived to our PCD clinic. It is possible that many other centers in the province don't know yet about our diagnostic PCD center and have not referred their patients to our care. In addition, as it is the first year of the study, we had relatively low number of patients (12 patients to date) and we had limited data from follow up visits. Furthermore, although our institute has a diagnostic center for PCD that perform all of the recommended diagnostic tests, many of the tests have limitations as mentioned above.
We present the first results of a provincial PCD registry in Canada. The demographic characteristics, clinical symptoms, diagnosis and management in our Alberta patients correlated with what was previously described in the literature. We found that even in this small cohort, there was still a high percentage of patients that were diagnosed later than 5 y of age, which can affect the progression of their disease. Even though the patients in our registry had multiple diagnostic modalities, the nasal NO and the genetic tests were most confirmative, suggesting their important role in the diagnostic process. Expanding the number of patients in the current provincial registry or in combination with other centers in Canada, may result in a few hundreds included patients, which will substantially strengthen the value of this work.
Our goal is to increase the number of patients in this registry by encouraging higher level of suspicion for PCD among physicians in Alberta and by a combined approach of diagnostic tests. We hope that this registry can serve as a model for other provinces in Canada to establish similar provincial registries followed by a national registry and probably joint international registries. These will allow a better networking of Canadian centers treating PCD patients and may improve standardization of diagnosis and management.
We thank all the children and families who participated in the study. This work was supported by a resident grant from the Woman and Children Health Research Institute to Dr. Cohn.
Dr. Cohn and Dr. Amirav conceived and wrote the first draft of the manuscript. Both authors revised and reviewed the manuscript.
Dr. Cohn and Dr. Amirav declare that they have no competing interests and did not receive any personal financial support.