Interstitial lung diseases, (ILD) or diffuse parenchymal lung diseases (DPLD) are umbrella terms used collectively to denote aetiologically heterogeneous group of disorders sharing clinical, radiographic and physiologic similarities. The treatment choices and prognosis vary substantially among the different causes of ILD and hence determining the correct etiology is of paramount importance. Connective tissue diseases are important causes of DPLDs and ILD is an important cause of morbidity and mortality in antisynthetase syndrome. The interstitial involvement in this increasingly recognized entity especially that with anti-PL-12 antibody positivity, tends to be severe and progressive with poor response to steroid therapy. We describe the case of a young lady with antisynthetase syndrome and diffuse glucocorticoid resistant non-specific interstitial pneumonia, who was salvaged and stabilized with a sequential combination therapy of cyclophosphamide and mycophenolate.
Antisynthetase syndrome, Interstitial lung disease, Anti-PL-12 antibody, Cyclophosphamide, Mycophenolate
The diffuse parenchymal lung diseases, often collectively referred to as the interstitial lung diseases (ILDs), include a group of disorders with variable etiology that are have in common certain similarities in clinical, radiographic, physiologic, or pathologic manifestations [1]. The term "interstitial" tends to suggest that the primary or predominant site of involvement is the pulmonary interstitium. The term is somewhat misleading, as many of these disease states are also associated with extensive involvement of alveoli and pulmonary vasculature. DPLDs may be divided into those that with known causes and those that are idiopathic. The most common identifiable causes of ILD are exposure to occupational and environmental agents [2] (inorganic or organic dusts), drug-induced pulmonary toxicity [3] and radiation-induced lung injury among others. ILD can also complicate the course of most of the connective tissue diseases (rheumatoid arthritis, systemic lupus erythematosus, scleroderma, mixed connective tissue disease, polymyositis/dermatomyositis, etc).
Interstitial lung disease (ILD) is known to complicate the course of idiopathic inflammatory myopathies and is a major cause of morbidity and mortality [4]. The pattern of interstitial involvement varies from nonspecific interstitial pneumonia, usual interstitial pneumonia, organizing pneumonia, and acute interstitial pneumonia. Antisynthetase syndrome (ASS) is a specific systemic autoimmune inflammatory myopathy presenting with arthritis or arthralgia, interstitial lung disease, fever, Raynaud's phenomenon, and mechanic's hands [5]. The syndrome is characterized by the presence of aminoacyl-transfer RNA synthetase autoantibodies (ASA). ILD in ASS can be rapidly progressive and have poor response to systemic glucocorticoids. We describe the case of a young lady with this syndrome who had progressive ILD despite standard immunosuppression and was salvaged with cyclophosphamide and mycophenolate mofetil therapy.
A 35-year-old lady, home maker, with no comorbid illnesses presented with 3 months symptoms of dry cough and progressive dyspnoea. She also complained of 3 kg weight loss over 3 months and itchy lesions on the extremities. She had no fever, allergic background, alteration of bowel/bladder habits or thyroid disease. She had generalized weakness and muscle pain. Physical evaluation revealed pigmented papules over the hands, legs and feet. Features of mechanics hands (Figure 1) were present. Chest auscultation revealed fine crackles over both lung bases. CT chest (Figure 2) revealed bilateral lower lobe ground glass opacities with some septal thickening and subpleural sparing, consistent with a non-specific interstitial pneumonia pattern. Blood counts, urinalysis, renal functions and thyroid tests were normal. ESR and CRP levels were elevated. 2D Echocardiogram revealed a normal left ventricular function and no evidence of PAH. Considering atypical pneumonia as one of the differentials, bronchoscopy with BAL evaluation for infective agents was done, all of which returned negative. Rheumatology consultation was requested for the skin lesions and arthralgia. Testing for ANA by immunoblot and myositis profile were done. ANA blot showed strong positivity for Ro 52 and Myositis profile showed strong positivity for PL-12 and Ro 52. A diagnosis of PL-12 related antisynthetase syndrome was made and she was counseled on potential disease course and prognosis. Our patient did not have clinical features of myositis, and muscle enzymes including CPK, LDH, SGOT and SGPT were within normal limits. So we did not proceed with electroneuromyography (ENMG). Her spirometry and DLCO showed moderate restriction and moderate limitation respectively (Figure 3a and Figure 3b). 6 MWT revealed exercise induced desaturation with a 6 MWD of 280 meters.
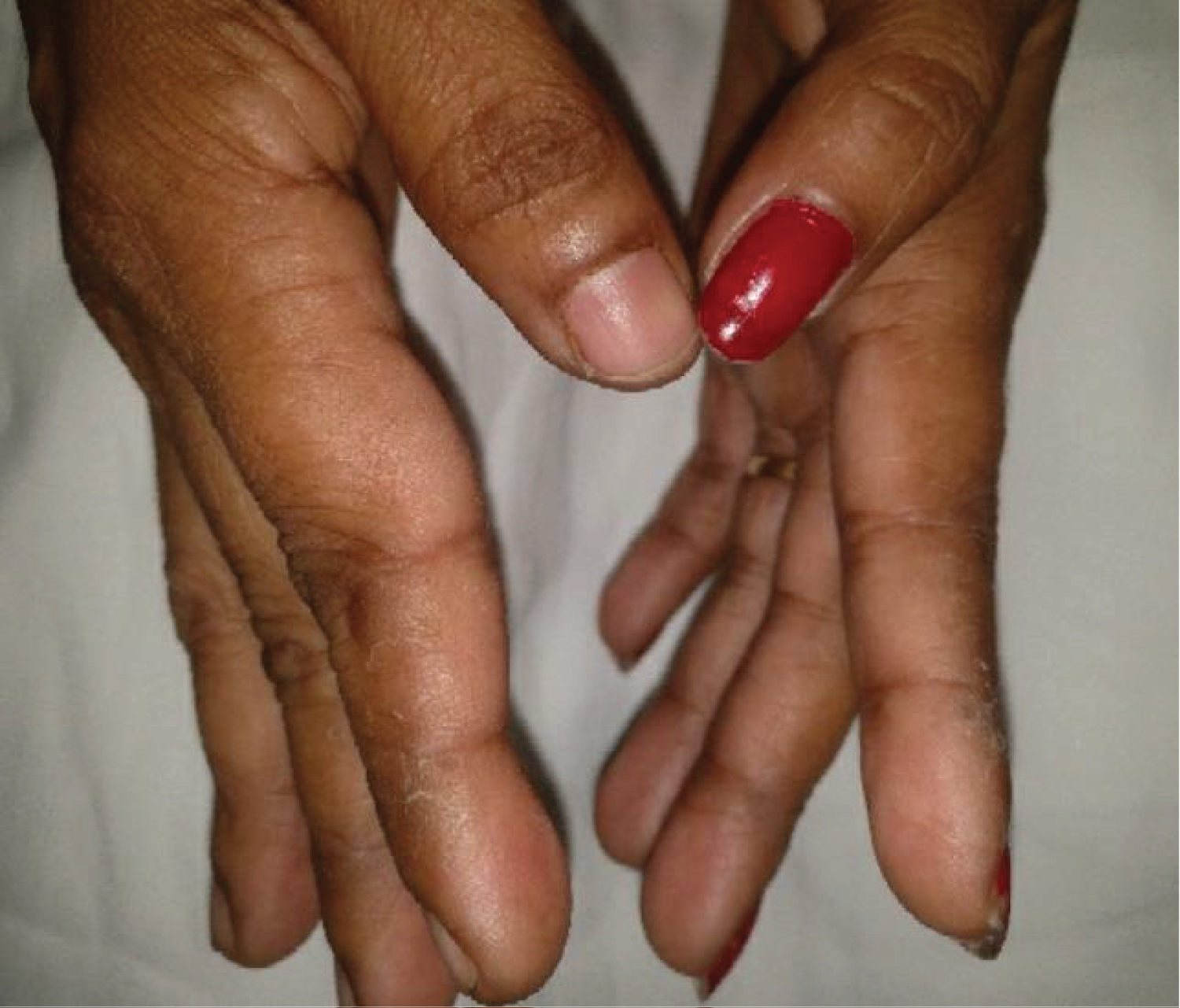 Figure 1: Mechanic's hand in patient with antisynthetase syndrome.
View Figure 1
Figure 1: Mechanic's hand in patient with antisynthetase syndrome.
View Figure 1
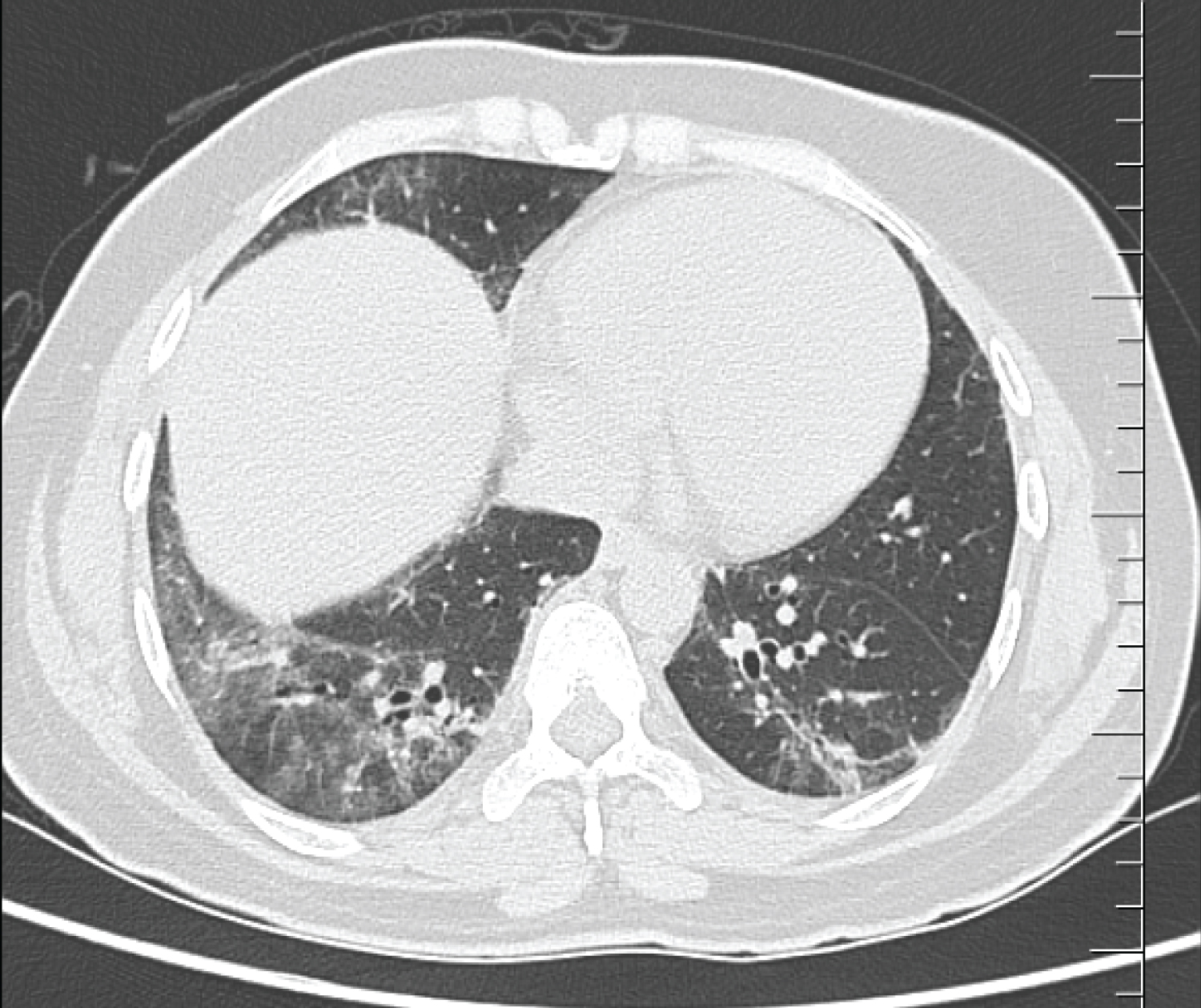 Figure 2: CT chest showing bilateral lower lobe ground glass opacities with some septal thickening and subpleural sparing, consistent with a non-specific interstitial pneumonia pattern.
View Figure 2
Figure 2: CT chest showing bilateral lower lobe ground glass opacities with some septal thickening and subpleural sparing, consistent with a non-specific interstitial pneumonia pattern.
View Figure 2
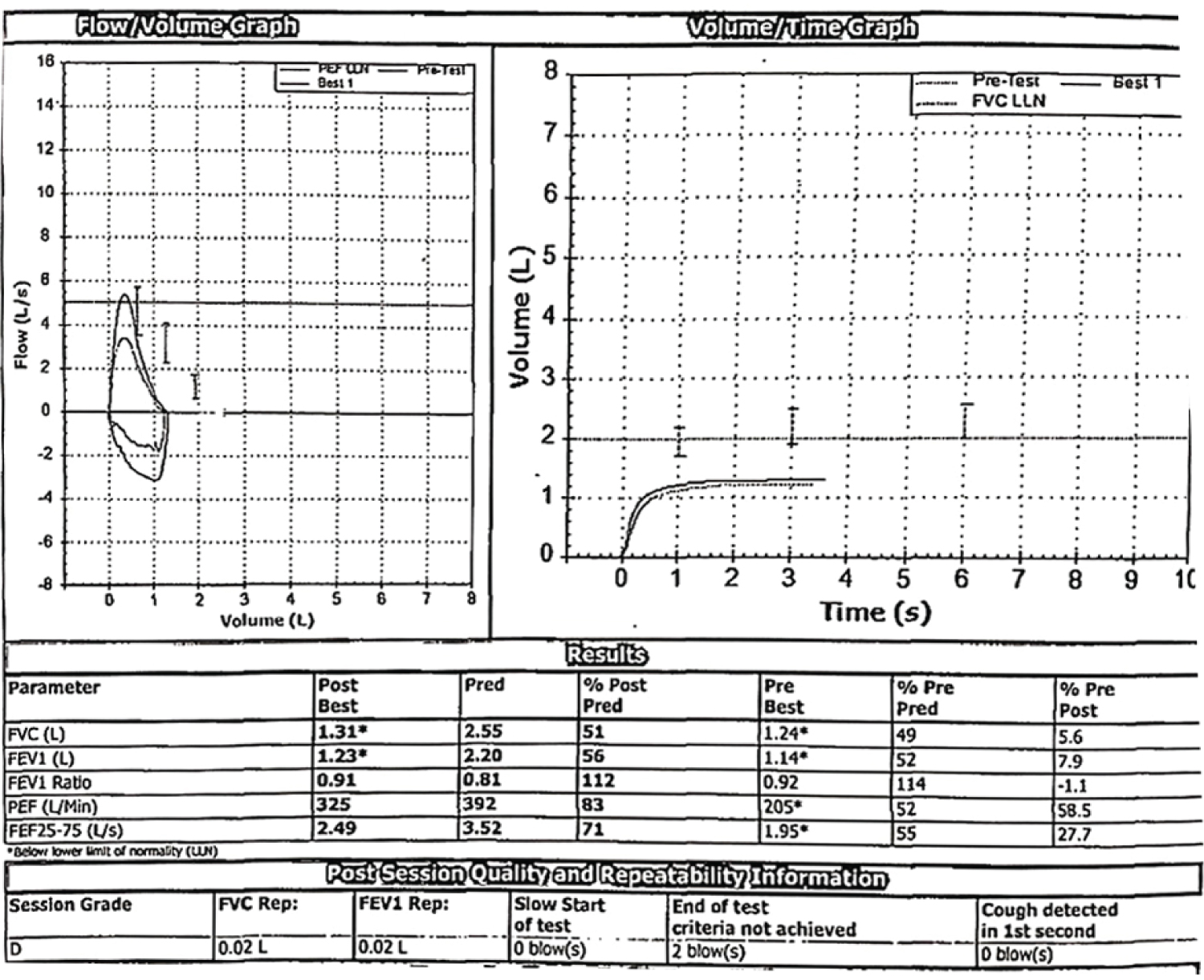 Figure 3a: Moderate restrictive ventilatory defect.
View Figure 3A
Figure 3a: Moderate restrictive ventilatory defect.
View Figure 3A
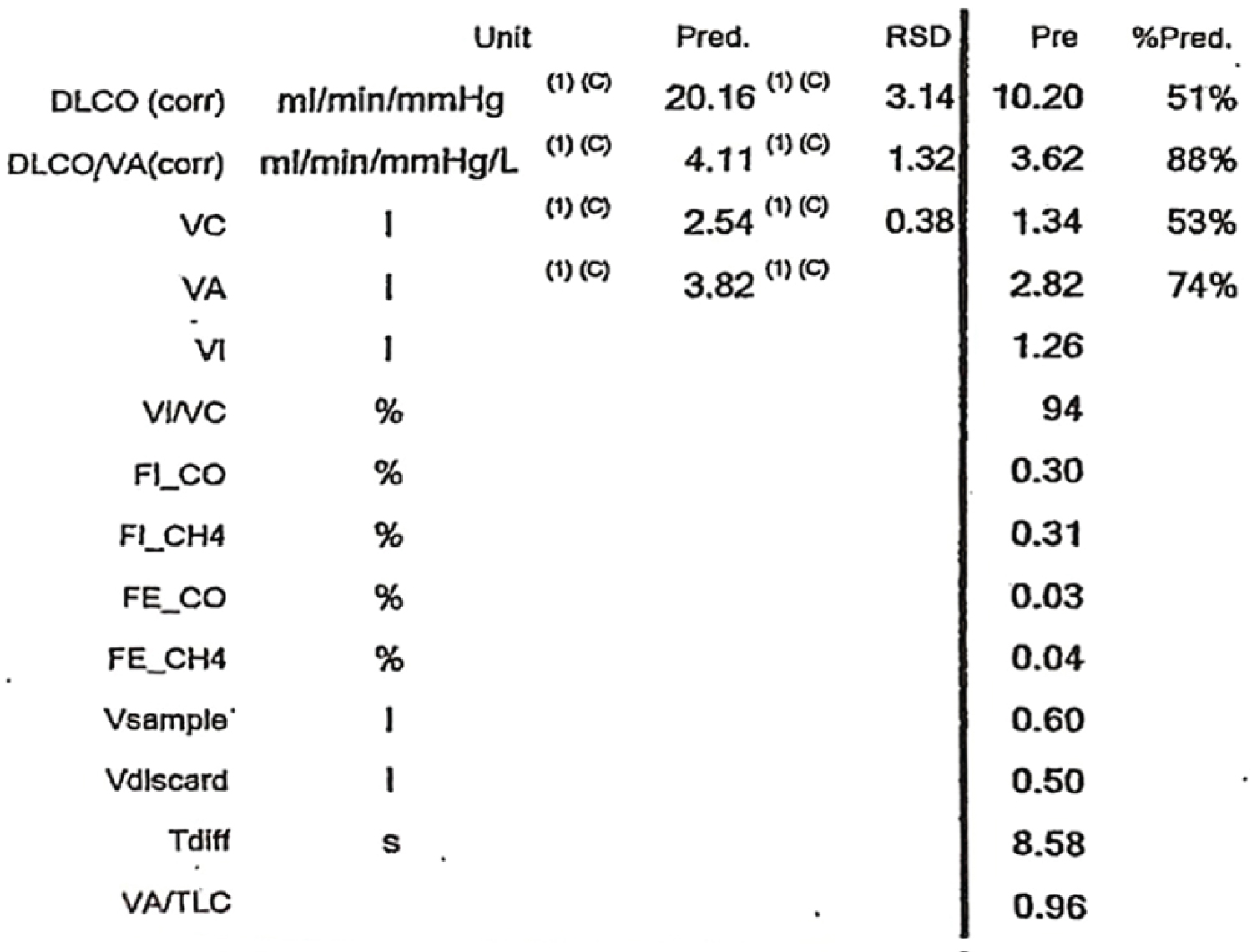 Figure 3b: Moderate limitation in diffusion.
View Figure 3B
Figure 3b: Moderate limitation in diffusion.
View Figure 3B
Pulse dose of methyl prednisolone was given (1 gram per day for 3 days) followed by prednisolone 60 mg per day in tapering doses. At 3 weeks of therapy, she remained symptomatic without clinical response. CT chest was repeated which revealed persisting ILD with worsening septal thickening. Spirometry revealed worsening FVC and a 6 MWD of 240 meters (fall of 40 meters from the baseline value). In view of the severe nature of the disease and lack of response to initial therapy, she was started on monthly Cyclophosphamide (1000 mg/m2) for 6 months. She improved symptomatically; lung function and radiological changes stabilized. Mycophenolate was added after 6 months (2 gram/day) and steroids were tapered. She remains on clinical, radiological and functional follow up with monitoring for any drug toxicity.
Idiopathic inflammatory myopathies (IIMs) are a heterogeneous group of acquired autoimmune diseases. Exhaustive reviews in this subject [5] suggest that based on their clinical and histopathological features, IIMs can be classified as polymyositis (PM), dermatomyositis (DM), inclusion body myositis and immune-mediated necrotizing myopathy. Myositis may also be associated with malignancy and can present as overlap syndromes with other connective tissue diseases. Antisynthetase syndrome is characterized by varying permutations of inflammatory myopathy, arthritis/arthralgia, ILD, fever, Raynaud's phenomenon, and mechanic's hands. It may be viewed upon as an entity overlapping with DM or PM and is associated with the presence of autoantibodies to aminoacyl-t RNA synthetase. These myositis specific autoantibodies are directed against cytoplasmic enzymes that catalyze the binding of specific amino acids to the matching tRNA during the translation phase of protein synthesis. There are currently eight identified antisynthetase autoantibodies of which the anti-Jo-1 (anti-histidyl-tRNA synthetase) and the PL-12 (anti-alanyl-tRNA-synthetase) account for majority of reported cases.
Dalakas and Holhfeld [6] proposed revised diagnostic criteria for idiopathic inflammatory myopathies in 2003, with a greater emphasis on histologic and immunologic pathology to distinguish between the IIM subtypes. Formal criteria for the diagnosis of anti-synthetase syndrome were introduced by Connors, et al. [7], which proposed that all patients with anti-synthetase syndrome must have evidence for a tRNA-synthetase autoantibody, in addition to one or more of the following clinical features: Mechanic's hands, Raynaud's phenomenon, myositis, ILD, arthritis, and/or unexplained fever. These criteria allowed for the recognition of the anti-synthetase syndrome as a distinct entity, which may be useful in both clinical and research settings.
Patients with ASS have features that overlap with other CTDs, such as Raynaud's phenomenon, severe gastroesophageal reflux disease, and non-erosive arthritis. Symptoms and signs of ILD predominate and are frequently the presenting feature. The prevalence of ILD in anti-synthetase syndrome is considered high. In a 2013 study [8] of 203 anti-synthetase syndrome patients, the prevalence of ILD was 86%, much more than the prevalence of myositis (73%) or arthralgia/arthritis (60%). In fact, anti-synthetase syndrome patients may not have classic myopathy symptoms at the initial stages and the myositis may present later in the disease course. The reported prevalence of autoantibodies in patients with an IIM varies widely. In a 2006 study of 74 patients [9] with IIM, anti-Jo-1 predominated, and was found to be prevalent in 30% (over half of all patients with myositis-specific antibodies were Jo-1 positive). A 2010 Australian study [10] of patients with anti-synthetase antibodies demonstrated a prevalence of the Jo-1 antibody in 88% of included patients. An Indian series of 9 patients with ASS associated ILD used anti Jo 1 antibody positivity as the diagnostic criteria [11].
Current knowledge classifies antisynthetase syndrome as a distinct entity within the group of inflammatory myopathies [12]. Immunosuppression is the cornerstone of treatment in anti-synthetase syndrome. Muscle and lung disease may not track together and require separate monitoring by an interdisciplinary team of rheumatologists and pulmonologists. Corticosteroids have long been first-line in the treatment of IIMs, though there is frequent lung disease recurrence with corticosteroid tapering. Additional immunosuppressive agents are added for refractory muscle and/or lung disease and as corticosteroid-sparing agents. Frequently used adjunctive agents include azathioprine, mycophenolate mofetil, tacrolimus, rituximab, and cyclophosphamide. Various combinations of immunosuppressive agents have been tried in patients with refractory ILD due to DM or PM. Glucocorticoids, monthly cyclophosphamide, and a calcineurin inhibitor may be used in combination [13]. Based on the limited data in support of this approach, an alternate choice would be one of the less well-established options, such as rituximab [14], intravenous immunoglobulin, and basiliximab.
Our report summarises the successful salvage story of a young lady with antisynthetase syndrome and ILD which was resistant to standard immunosuppressive therapy resistant ILD. She was salvaged with sequential addition of cyclophosphamide and then mycophenolate. The case is noteworthy on multiple accounts. Antisynthetase syndrome as a cause of rapidly progressive ILD is increasingly recognized as in the present case. Most reported case series from India have anti Jo-1 related ASS, as opposed to anti-PL-12 antibody, substantiating the need of testing for an extended myositis antibody panel is suspected cases of ILD due to ASS. The importance of continued clinical, radiological and functional monitoring (with lung function tests) to ensure prompt response cannot be over stressed and salvage therapy with multiple agents may be needed in non-responders.
Nil.
Dr. Anand V, Dr. Chandrasekhar S, Dr. Aparna S Nirmal, Dr. Fathima KM, Residents in the Department of Pulmonary Medicine.