The development of cell lines with high cell density, controlled proliferation, apoptosisresistance, and easy adaptation into cultures of serum free media is essential for the success of tissue engineering strategies. This study aimed to examine the effects of co-overexpression of c-myc and bcl-2 on viability, apoptosis and differentiation of Dental Pulp Stem Cells (DPSCs). DPSCs were transduced with premade lentiviral particles of human target c-Myc and bcl-2 consecutively. Western blot results confirmed the over-expression of c-Myc and Bcl-2 in respective cell lines. c-Myc over-expressing DPSC cultures showed an increase in proliferation rate and maximum cell number compared to wild type DPSCs as shown by cell counting kit-8 assay. However, c-Myc overexpression also demonstrated significantly higher apoptotic rates in DPSCs than that of wild type DPSCs when cultured under serum starvation. c-Myc overexpressing DPSCs co-transduced with bcl-2 resulted in reduction of apoptosis levels as shown by cell death detection ELISA kit and caspase-3 colorimetric assay, without affecting the cell proliferation rate. Further, c-Myc overexpression appeared to reduce osteo/odontogenic differentiation capacity, whereas Bcl-2 co-overexpression demonstrated a slight increase in differentiation levels. c-Myc and Bcl-2 co-overexpression increases the cell viability in cell proliferation and reduce apoptosis and cell death in DPSCs.
Apoptosis, Bcl-2, Cell viability, c-Myc, Dental Pulp Stem Cells (DPSCs), Gene modification
Dental Pulp Stem Cells (DPSCs), which could be obtained in large pools of autologous cells, is considered as a promising population of stem cells in regenerative medicine [1]. However, limited self-renewal and proliferation capacity restrict the applications of DPSCs in clinical setting [2]. Therefore, modification of cell lines to achieve high cell density, enhanced proliferation, apoptosis resistance, and easy adaptation into cultures of serum free media is essential for the success of tissue engineering strategies [3].
In tissue engineering, optimization of methods in cell culture techniques is a progressing arena as the enhanced knowledge of the complex dynamics in regulation of cell survival and growth contributes to improve the expected outcome. Cell engineering strategies to improve maximal cell number would be a major step towards reaching the goal of optimally engineered tissue constructs. Overexpression of important regulators such as growth factors and cell cycle genes of proliferation and apoptosis pathways is one of the strategies employed in cell culture for the management of cell proliferation [4].
The use of proto-oncogenes to modulate the cell proliferation is a potential solution in enhancing the capacity of DPSCs' use in regenerative medicine [5]. Overexpression of c-myc, a transcription factor that is involved in regulation of cell cycle, has been shown of capable in inducing cell proliferation [6,7]. The c-myc encodes a nuclear phosphoprotein, which after binding to DNA, plays an essential role in transcriptional activation of target genes, as well as in proliferation and apoptosis [8]. However, c-Myc not only plays an important role in cell proliferation but also acts in induction of apoptosis [8]. Therefore, it appears that cell proliferation and apoptosis are coupled at least when it is under c-Myc regulation [9]. Bcl-2 that is involved in cell survival is a prime candidate to be introduced in cell lines to regulate the apoptosis induced by c-myc [10,11]. It was shown that Bcl-2 expression effectively inhibits c-Myc-induced apoptosis without affecting the c-Myc mitogenic function [6,8]. This synergistic interaction between c-Myc and Bcl-2 may have implications for developing cell lines with enhanced potential in tissue engineering.
If the aforementioned properties of c-Myc and Bcl-2 expression could be achieved in target progenitor cells, it could lead to development of cell lines with apoptosis resistance, high cell density, controlled proliferation and easy adaptation into serum free medium. Therefore, in the present study, we aimed to overexpress c-myc and bcl-2 in the DPSCs using premade lentiviral particle transfection technique and examine its effects on DPSCs' viability in cell proliferation, survival and odonto/osteogenic differentiation capacity.
Premade human target, bcl-2, c-myc lentiviral particles and respective null-control particles were commercially purchased from GenTarget Inc, San Diego, CA. DPSCs were commercially purchased from All Cells, LLS., Alameda, CA and cultured in α-Minimum Essential Medium (α-MEM) supplemented with 15% fetal bovine serum, L-ascorbic acid-2-phosphate, 100 U/mL penicillin-G, and 100 mg/mL streptomycin under 5% CO2 at 37 °C. Passage 3 to 6 cells of each cell type were used in all experiments with the parameters described later.
DPSCs were transfected for the expression of c-Myc-RFP or both c-Myc-RFP and Bcl-2-GFP genes as described by the manufacturer. Briefly, DPSCs of passage 3 were seeded at 0.5 × 105/ml 0.5 ml in a well of a 24-well plate and incubated overnight. At 70% confluence, the culture medium was replaced with 0.5 ml fresh, complete medium and 50 μl lentivirus stock was added. After incubating the cultures for 72 hours in a 37 °C/CO2 incubator, transfection rate was checked under fluorescence microscope. c-Myc-RFP, Null-RFP, c-myc-RFP-Bcl-2-GFP and Null-RFP-GFP stably over-expressing cell lines were selected using puromycin or/and blastomycin antibiotics. The antibiotic's kill curve for DPSCs was determined in a pilot experiment.
The whole-cell lysate was obtained using M-PER Mammalian Protein Extraction Reagent (Thermo Scientific, Rockford, IL, USA) and quantified with a BCA protein assay kit (Thermo Scientific). Then, 30 mg of protein was subjected to 12% SDS-PAGE (Bio-Rad Laboratories) and blotted onto polyvinylidene fluoride membranes (GE Healthcare, Uppsala, Sweden). After blocking for 1 h in TBS-T (20 mM Tris-HCl, 150 mM NaCl, 0.05% Tween-20) containing 5% skim milk, the membranes were washed three times in TBS-T and incubated with specific anti-c-Myc and anti-Bcl-2 antibodies (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) overnight at 4 °C. Following three washes in TBS-T, the membranes were probed with second HRP-conjugated anti-rabbit Ab for 2 h. The membranes were developed using enhanced chemiluminescence reagents (Thermo Scientific). The proteins were normalized to β-actin (Santa Cruz Biotechnology, Inc.) levels. The density of bands was analyzed by Quantity One 4.6.9 software (Bio-Rad Laboratories).
Different groups of DPSCs were inoculated in a 96-well plate at a concentration of 104 cells per well and incubated in a 37 °C, 5% CO2 humidified incubator. Ten microliters of the CCK-8 solution (Sigma-Aldrich, Inc., St. Louis, MO) was added to each well of the plate. After incubating the plate for 4 hours in the incubator, the absorbance at 450 nm was measured using a microplate reader.
Different groups of DPSCs were added to a 48-well plate at a concentration of 104 cells per well and cultured with or without serum in a 37 °C, 5% CO2 humidified incubator. Briefly, at 80-90% confluence, the cells were starved with serum-free medium for 12 hours and at various timepoints, cell lysates were harvested for subsequent analyses. Cell apoptosis was measured using Cell Death Detection ELISAPlus Kit (Roche Diagnostics GmbH, Mannheim, Germany) according to the manufacturer's protocol. Briefly, at each time point, 200 μl of lysis buffer was added to each well and incubated for 30 minutes at room temperature. The lysate was centrifuged at 200× g for 10 minutes and 20 μl from the supernatant was transferred to the streptavidin coated microplate wells for analysis. After adding 80 μl of the immunoreagent (mixture of Anti-histone-biotin and Anti-DNA-POD) that detects nucleosomes in the samples to each well, the microplate was covered with an adhesive cover foil and incubated on a microplate shaker under gently shaking for 2 hours at room temperature. Solutions were removed thoroughly by tapping and each well was rinsed three times with 300 μl incubation buffer. After pipetting 100 μl of ABTS solution to each well, the microplate was incubated on a plate shaker at 250 rpm for 20 minutes. The reaction was stopped by adding 100 μl of ABTS stop solution and the absorbance at 405 nm was measured using a microplate reader. The well with only ABTS solution and ABTS stop solution was measured as a blank.
Cells were cultured in a similar manner to that described above under cell apoptosis assay. Caspase 3 activity of the different groups of DPSCs was measured using Caspase-3 Colorimetric Assay kit (R&D Systems, Inc., Minneapolis, MN, USA). Briefly, whole-cell lysate was collected adding the Lysis Buffer. After incubating on ice for 10 minutes, the cell lysate was centrifuged and the supernate was transferred to a new tube. Fifty microliters of cell lysate, 50 μl of 2 × Reaction Buffer and 5 μl of Caspase-3 colorimetric substrate were added to each reaction well of a microplate and incubated at 37 °C for 2 hours. The plate was read on a microplate reader using a wavelength of 405 nm. No cell lysate and no substrate groups were included as controls of the assay.
Cells of different groups of DPSCs were seeded on 6-well plates at a concentration of 1 × 105 cells per well. Odonto/osteogenic differentiation of cultures was induced at 70% cell confluence by medium supplemented with 10 nmol/L dexamethasone, 10 mmol/L β-glycerophosphate, and 50 mg/mL L-ascorbic acid phosphate up to 14 and 21 days for Alkaline Phosphatase and Alizarin Red assays, respectively.
ALP activity of different groups of DPSCs was assessed using the Sensolyte pNPP Alkaline Phosphatase Assay Kit - Colorimetric (AnaSpec, Inc., Fermont, CA). The cultures were rinsed twice with 1X assay buffer, solubilized in Triton X-100 in assay buffer, and scraped off the adherent cells. After incubating the scraping at 4 °C for 10 min under agitation, the suspension was centrifuged at 2500× g for 10 minutes at 4 °C. The supernatant was collected and used to detect the alkaline phosphatase (ALP) levels. Fifty microliters of samples and serially diluted ALP standard solutions from 200 to 0 ng/ml were added to the wells in a 96-well plate. Aliquot (50 μl) of freshly prepared p-nitrophenyl-phosphate substrate solution was added into each well and mixed the reagents by gently shaking the plate for 30 seconds. The reaction was incubated for 30 minutes at 37 °C and the optical density of p-nitrophenol at 405 nm was measured with spectrophotometry. ALP activity was presented as nanograms of p-Nitrophenyl phosphate per milliliter.
The DPSC cultures induced for 21 days were fixed with 10% formaldehyde at room temperature for 15 minutes and were washed with distilled water for 3 times. Alizarin Red solution was added and incubated at room temperature for 20 minutes. After washing 4 times with deionized water, 1 mL of water was added to each well to prevent the cells from drying, and the images were captured.
The quantification of Alizarin Red stain absorbed by the mineralized nodules was carried out as follows. A solvent of 10% acetic acid was added to each well, incubated for 30 minutes and the slurry was collected into a tube. The tube was vortexed for 30 seconds and heated to 85 for 10 minutes. After cooling, the slurry was centrifuged at 20,000 g for 15 minutes, and the supernatant was collected. The standard/sample was added to an opaque-walled, transparent-bottom 96-well plate, and optical density was measured at 405 nm with spectrophotometry.
Total RNA from cultures of different cell groups and time points were extracted with the RNeasy Mini Kit (Qiagen, Hilden, Germany). A 1.0 μg sample of total RNA was used to synthesize complementary DNA by SuperScript III Reverse Transcriptase (Invitrogen, Carlsbad, CA). The resultant complementary DNA concentrations were quantified using a NanoDrop ND-1000 Spectophotometer. Quantitative real-time Polymerase Chain Reaction (PCR) was performed using the ABI Prism 7000 Sequence Detection System (Applied Biosystems, Carlsbad, CA) with SYBR green (Applied Biosystems). All samples were run in triplicate in 96-well plates, with each well containing 1.20 mL complementary DNA diluted for a total reaction volume of 20 mL. Reactions were performed at 95 °C for 10 minutes followed by 40 cycles of 95 °C for 15 seconds and 60 °C for 1 minute. Primers for the differentiation markers ALP, Bone Sialoprotein (BSP), Dentin Sialophosphoprotein (DSPP), Runt-Related Transcription Factor 2 (RUNX2), Dentin Matrix Acidic Phosphoprotein 1 (DMP1) and the housekeeping gene Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) were designed using Primer Express software from Applied Biosciences. Primer sequences used in PCR are shown in Table 1. For data analysis, the Step One software v2.0.2 (Applied Biosystems) calculated the levels of target gene expression in samples relative to the level of expression in the control (non-induced) samples with the comparative cycle threshold method (∆∆CT). Expression values for target genes were normalized to the expression of GAPDH.
Table 1: The human primer sequences used for Real-Time PCR. View Table 1
The proliferating rate, apoptotic level, caspase-3 activity, ALP concentration and Alizarin Red concentration were expressed as a median ± quartiles and were statistically analysed using nonparametric statistical test Kruskal-Wallis one-way analysis of variance. All the experiments were conducted in triplicate. P < 0.05 was considered statistically significant.
To assay the effects of overexpression of c-Myc and Bcl-2 in DPSCs, the cells were transfected with target premade lentiviral particles. Observation under fluorescence microscope confirmed the successful transfection of cells with the expression of green and red fluorescent proteins. Puromycin or/and blastomycin resistant colonies were selected as the stably transfected cells. Western blot results confirmed the overexpression of c-Myc and Bcl-2 proteins in respective cell lines (Figure 1).
 Figure 1: Western blot analysis of c-Myc and Bcl-2 overexpression in different groups of DPSCs 1 week after transduction with lentiviral particles.
View Figure 1
Figure 1: Western blot analysis of c-Myc and Bcl-2 overexpression in different groups of DPSCs 1 week after transduction with lentiviral particles.
View Figure 1
c-Myc over-expressing DPSC cultures showed an increase in proliferation rate and maximum cell number compared to the wild type cells as shown by cck-8 assay (Figure 2). On the day 7 of culture, both c-Myc and c-Myc-Bcl-2 co-overexpressing DPSCs demonstrated a significantly higher (p < 0.05) cell proliferation than that of wild type DPSCs.
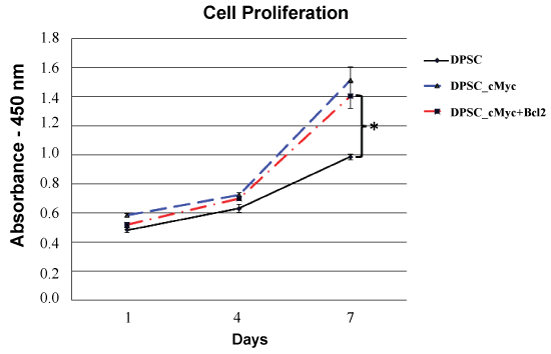 Figure 2: Cell proliferation rates of different groups of DPSCs as shown by CCK-8 assay. Both c-Myc overexpressing and c-Myc + Bcl-2 co-overexpressing cells showed significantly increased proliferation rates compared to wild type DPSCs (*p < 0.05).
View Figure 2
Figure 2: Cell proliferation rates of different groups of DPSCs as shown by CCK-8 assay. Both c-Myc overexpressing and c-Myc + Bcl-2 co-overexpressing cells showed significantly increased proliferation rates compared to wild type DPSCs (*p < 0.05).
View Figure 2
The pattern of apoptosis was established using Cell Death Detection ELISA kit and Caspase-3 Colorimetric assay. Figure 3 shows the levels of apoptotic death in the cultures of c-Myc overexpressing DPSCs with or without co-overexpression of Bcl-2 in comparison with wild type DPSCs in the presence of serum. Apoptosis in all three culture groups increased with time at a nearly constant rate and remained low even on day 7. However, c-Myc overexpressing DPSCs still showed a higher level of apoptosis compared with that of Bcl-2 co-overexpressing and wild type DPSCs.
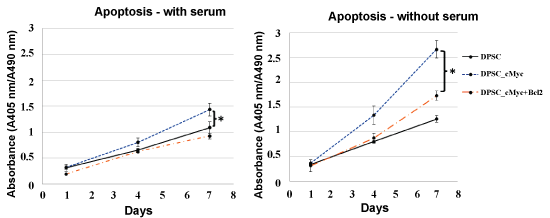 Figure 3: Apoptosis in different cultures of DPSCs in the presence and absence of serum. c-Myc alone overexpressing DPSCs demonstrated significantly higher apoptotic levels compared to wild type cells when cultured under serum starvation (*p < 0.05). c-Myc and Bcl-2 co-overexpressing cells showed comparable level of apoptosis to that of wild type DPSCs.
View Figure 3
Figure 3: Apoptosis in different cultures of DPSCs in the presence and absence of serum. c-Myc alone overexpressing DPSCs demonstrated significantly higher apoptotic levels compared to wild type cells when cultured under serum starvation (*p < 0.05). c-Myc and Bcl-2 co-overexpressing cells showed comparable level of apoptosis to that of wild type DPSCs.
View Figure 3
In the absence of serum, the difference in the levels of apoptosis between c-Myc overexpressing cells and other groups was significant from day 4 of the culture, with c-Myc overexpressing DPSCs showing significantly higher apoptotic rates than wild type and Bcl-2 co-overexpressing DPSCs (p < 0.05). Caspase-3 activity, which is an important marker of apoptosis further confirmed a similar pattern in different groups of DPSCs (Figure 4).
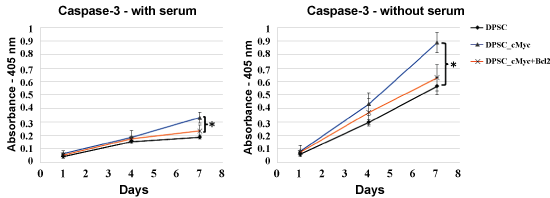 Figure 4: Caspase-3 activity in different cultures of DPSCs in the presence of serum and absence of serum. Caspase-3 levels were significantly higher in c-Myc alone overexpressing DPSCs compared to the c-Myc and Bcl-2 co-overexpressing and wild type cells (*p < 0.05).
View Figure 4
Figure 4: Caspase-3 activity in different cultures of DPSCs in the presence of serum and absence of serum. Caspase-3 levels were significantly higher in c-Myc alone overexpressing DPSCs compared to the c-Myc and Bcl-2 co-overexpressing and wild type cells (*p < 0.05).
View Figure 4
Bcl-2 co-overexpressing cells demonstrated a lower apoptosis and higher cell survival compared to the c-Myc overexpressing DPSCs. Over-expression of Bcl-2 in c-Myc overexpressing cells reduced the apoptosis while not compromising their proliferation capacity. This resulted in a cell line that exhibited higher proliferation rates and maximum cell numbers, with a decrease in apoptosis when compared to the parental cell line.
To investigate whether oncogene overexpressing DPSCs retain a comparable level of odonto/osteogenic differentiation capacity to their parental DPSCs, ALP activity and mineralization were quantified in induced cultures. Results showed that c-Myc overexpressing DPSCs have a reduced level of ALP activity than that of wild type DPSCs. However, c-Myc and Bcl-2 co-overexpression results in increased ALP activity compared to c-Myc alone overexpression (Figure 5).
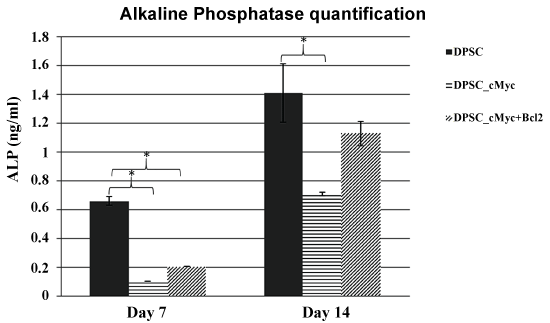 Figure 5: ALP activity in different cultures of DPSCs induced for odonto/osteogenic differentiation. c-Myc overexpressing DPSCs showed decreased levels of ALP activity in comparison to wild type DPSCs (*p < 0.05).
View Figure 5
Figure 5: ALP activity in different cultures of DPSCs induced for odonto/osteogenic differentiation. c-Myc overexpressing DPSCs showed decreased levels of ALP activity in comparison to wild type DPSCs (*p < 0.05).
View Figure 5
Alizarin red staining of the 21-days induced cultures revealed calcified nodules in the extracellular matrix (Figure 6A). The wild type DPSC cultures showed more pronounced calcifications compared with c-Myc overexpressing DPSC cultures with or without Bcl-2 co-overexpression as shown by Alizarin Red staining and quantification (Figure 6B) of the stains absorbed by the cultures. These results suggested that c-Myc overexpression delays the odonto/osteogenic differentiation process of DPSCs.
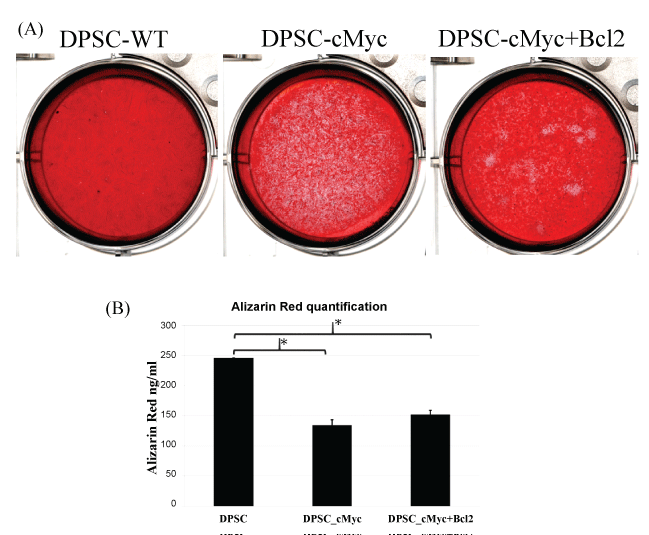 Figure 6: Alizarin Red. A) Staining and; B) Quantification of mineralization in different cultures of DPSCs after 21 days of induction for odonto/osteogenic differentiation. Mineralization was decreased in c-Myc overexpressing DPSCs compared to wild type DPSCs (*p < 0.05).
View Figure 6
Figure 6: Alizarin Red. A) Staining and; B) Quantification of mineralization in different cultures of DPSCs after 21 days of induction for odonto/osteogenic differentiation. Mineralization was decreased in c-Myc overexpressing DPSCs compared to wild type DPSCs (*p < 0.05).
View Figure 6
mRNA expression levels of DSPP, DMP1, ALP, BSP, and RUNX2 genes in different groups further confirmed the relatively reduced odonto/osteogenic differentiation in c-Myc overexpressing DPSCs (Figure 7). In accordance with other results, c-Myc Bcl-2 co-overexpressing DPSCs expressed higher levels of odonto/osteogenic marker expression compared to c-Myc alone overexpressing cells (Figure 7).
 Figure 7: mRNA expression levels of DSPP, DMP1, ALP, BSP, and RUNX2 genes in different cultures of DPSCs. All the examined differentiation marker levels were relatively reduced in c-Myc overexpressing cells compared to wild type DPSCs. c-Myc and Bcl-2 co-overexpressing cells demonstrated a slightly enhanced expression levels in comparison to that of c-Myc alone overexpressing DPSCs (*p < 0.05).
View Figure 7
Figure 7: mRNA expression levels of DSPP, DMP1, ALP, BSP, and RUNX2 genes in different cultures of DPSCs. All the examined differentiation marker levels were relatively reduced in c-Myc overexpressing cells compared to wild type DPSCs. c-Myc and Bcl-2 co-overexpressing cells demonstrated a slightly enhanced expression levels in comparison to that of c-Myc alone overexpressing DPSCs (*p < 0.05).
View Figure 7
In this report we show that overexpression of c-Myc protein can increase proliferation of DPSCs and co-expression of Bcl-2 can counter-act the apoptosis induced by c-Myc under serum free conditions. Bcl-2 and c-Myc are two major oncogenic proteins that can act together in cell proliferation, transformation, and apoptosis [11]. c-Myc is a transcription factor that promotes cell growth by regulating the expression of genes that control the G1 to S phase transition. Overexpression of c-Myc could force quiescent cells into S phase in the absence of required mitogens and prevents withdrawal of cells from the cell cycle [12]. However, c-Myc sensitizes cells to apoptosis under certain conditions, including cytokine deprivation, chemotherapeutics, DNA damage, hypoxia, heat shock, TNF-α, and antigen receptor activation [13].
Previous studies have clearly shown that high c-Myc protein levels do not essentially make a cell vulnerable to enhanced apoptosis [9]. Those reports suggest that c-Myc functions as a permissive factor rather than an initiating factor of apoptosis.
In contrast, Bcl-2 is a proto-oncogene that is known for its anti-apoptotic properties. There is convincing evidence that the expression of Bcl-2 can prolong the life of cells that would otherwise die of apoptosis. It was further shown that Bcl-2 functions as one of the most potent c-Myc cooperating oncoproteins and acts to avoid c-Myc-induced cell death ensuring its mitogenic property [6,8,12].
Therefore, we chose to overexpress c-Myc and Bcl-2 in primary DPSCs to examine their effects on proliferation and survival of DPSCs. Similar to the results of previous studies, expression of high levels of Bcl-2 prolonged the survival of DPSCs in the absence of serum; in addition, Bcl-2 expression blocked c-Myc-induced apoptosis in the absence of serum.
Induction of apoptosis by c-Myc and inhibition by Bcl-2 were distinctly observable by 4 and 7 day cultures of serum starvation, and protection by Bcl-2 persisted for at least for 1 week. Although Bcl-2 is known for its anti-apoptotic properties how Bcl-2 actually inhibits apoptosis is unclear. Wagner, et al. reported that the protective effect of Bcl-2 is not a result of altering cell cycle progression since they did not observe any change in cell cycle profile attributable to expression of Bcl-2 [10]. Furthermore, some other studies have reported that Bcl-2 expression has no effect on cell cycle progression in pro-B cells [14] and does not block establishment of quiescence in NIH3T3 cells in the absence of serum [15]. The ability of Bcl-2 expression to repress c-Myc-induced apoptosis in serum-starved DPSCs suggests that this form of apoptosis shares a common mechanism with apoptosis induced in cell lines as described above.
Bcl-2 is shown to be expressed following mitogenic activation of peripheral blood lymphocytes [16], although it is generally expressed constitutively throughout the cell cycle [17]. In addition, its localization to the inner mitochondrial membrane [14] suggests a potential role as a regulator of intracellular Ca2+ concentration, since Ca2+ acts both as a second messenger in the signal pathway from growth factor receptors to the nucleus and as an inducer of apoptosis [18].
Differentiation and proliferation are generally mutually exclusive events, and when the proliferation is enhanced, there is a probability for the differentiation to be affected. It was shown that c-Myc expression is rapidly downregulated in many cell types during differentiation [19,20]. Series of antisense experiments have demonstrated that the downregulation of c-Myc is crucial for differentiation to proceed [19,21,22]. Conversely, c-Myc overexpression was shown to inhibit differentiation in a number of cell types [19,20]. Accordingly, the results of the present study also showed a lower differentiation capacity in c-Myc overexpressing DPSCs compared with that of parental DPSCs. However, c-Myc and Bcl-2 co-overexpressing DPSCs demonstrated a higher differentiation capacity than that of c-Myc overexpressing DPSCs. Therefore, in addition to its anti-apoptotic effect, bcl-2 expression may have an inhibitory effect on c-Myc's anti-differentiation function. The mechanism (s) by which c-Myc blocks cellular differentiation, however, remains poorly defined [19,20].
Although it seems reasonable to propose that the proliferative function of c-Myc and anti-apoptotic function of Bcl-2 could be used in enhancing the therapeutic potential of DPSCs in tissue engineering and regenerative medicine, the inappropriate expression of Bcl-2 or/and c-Myc appears to play a role in the immortalization of certain malignant cells. Therefore, further studies are required to examine the long-term effects of the c-Myc and Bcl-2 overexpression of benign primary cells prior to use them in clinical applications.
None.
This work was supported by grants from National Nature Science Foundation of China (81271135) to C. Zhang and Small Project Funding (201409176177) to WL Dissanayaka.