Alport syndrome is an inherited renal disease characterized by haematuria, renal failure and hearing loss. Factors that determine progression to ESKD are not well described and it is uncertain whether time to ESKD and mortality have improved over time.
In this Irish national retrospective case series we describe the clinical features and outcomes of all patients with Alport syndrome diagnosed over the past 40 years. Our aim was to examine the factors that predict time to ESKD and patient mortality and to assess how these factors may have varied over time. Patients were divided into an older (1974-1994) and a more modern era (1995-2015) according to their date of diagnosis.
133 patients were diagnosed with Alport syndrome between 1974 and 2015. 59% of patients reached ESKD during the period of study. 10-year ESKD-free rate was 53.7% (95% C.I. 31.6-71.5%) for the earlier era, compared to 60.8% (95% C.I. 41.5-75.5%) for the later era. Female sex was predictive of reduced ESKD risk as was lower serum creatinine at diagnosis. 10-year patient survival was 92.3% (95% C.I. 72.5-98.0%) in the first era compared to 97.1% (95% C.I. 80.9-99.6%) in the second. There was no effect of era on patient outcome.
There is no difference in time to ESKD and in overall patient survival for Alport patients between the older and modern era, though patient survival is still significantly better than that of patients with non-Alport renal disease.
End-stage kidney disease, Alport syndrome, Chronic kidney disease, Hereditary nephritis, Dialysis, Disease progression
Alport syndrome is an inherited renal disease, first described in 1927 [1], which is characterized by haematuria, renal failure, hearing loss, lenticonus, retinal flecks [2], a lamellated glomerular basement membrane [3] and mutations in the COL4A5 or COL4A3/COL4A4 genes [4] leading to abnormal Type IV collagen composition [5]. The prevalence of the disease is estimated at 1 in 50,000 live births [6]. Eighty-five percent of families have X-linked inheritance with mutations in COL4A5 [7]. Most of the others have autosomal recessive inheritance, with homozygous or compound heterozygous mutations in both gene copies of COL4A3 or COL4A4 [8]. Autosomal dominant inheritance is rare and results from heterozygous COL4A3 or COL4A4 variants [9].
Males with X-linked disease express a more severe phenotype than females [10]. Autosomal recessive inheritance is suspected when disease occurs sporadically in just one generation or within a consanguineous family. Males and females within the family are affected with the same frequency and severity. The father may also be affected by haematuria and females more frequently suffer from renal failure, hearing loss and ocular abnormalities.
Proteinuria, hearing loss, lenticonus, retinopathy and reduced levels of Type IV α5 chain GBM collagen all correlate with an increased risk of early onset renal failure in males, but the risks have not been studied prospectively [11]. Almost all females with X-linked Alport syndrome have haematuria (95%) and many others develop further clinical features including proteinuria (75%) [12], progression to end-stage kidney failure (8-30%), hearing loss (40%) and peripheral retinopathy (40%) [13]. X-chromosome inactivation (lyonization) is protective for females in preventing damage to the collagen network and leading to less penetrance of clinical features.
Patients with Alport syndrome who progress to end-stage kidney disease usually do so at a young age, with men typically commencing renal replacement therapy a decade before their female counterparts [14]. Mallett, et al. [15] recently noted that factors predicting time to ESKD and outcomes of same have not been well-studied to date. They found that Alport syndrome is overall an uncommon cause of ESKD and that it is associated with younger age at commencement of renal replacement therapy, male gender, earlier referral and a higher likelihood of receiving a renal transplant.
In this large national retrospective case series, based in Beaumont Hospital Dublin, we describe the clinical features and outcomes of all patients with biopsy-proven or clinically suspected Alport syndrome diagnosed and managed there over the past 40 years. A systematic search of histopathology records and of the EMedRenal Clinical and Patient Information Software System, which is used nationally to integrate renal patient data for clinical and statistical purposes. Our aim was to examine the factors that help predict time to ESKD and patient mortality and to assess how these factors may have varied over time. For the purpose of comparison, patients were divided into an older (1974-1994) and a more modern era (1995-2015) according to their date of diagnosis/renal biopsy, in order to assess for a significant difference in patient outcomes over time.
Descriptive features were presented using frequencies and percentages for categorical variables and either mean and standard deviation or median and interquartile range for continuous variables. Fisher Exact, T test or Wilcoxon Rank sum tests were used to compare the eras according to data distribution. The main outcomes that were assessed were patient survival and time to end-stage kidney disease from time of diagnosis.
Survivor functions were derived using the Kaplan-Meier method for the time to event graphs. Cox Proportional Hazards models were performed to assess the effect of era on outcome in the presence of potential confounding variables. Statistical analysis was performed using Stata SE Version 13 software (College Station, Texas). The probability of a type 1 error of less than 5% was used to determine significance.
One hundred and thirty-three patients were diagnosed with Alport syndrome in Beaumont Hospital between 1974 and 2015. The primary method of diagnosis was by renal biopsy. 107/133 (80%) patients in total underwent native renal biopsies. 45/107 (42%) patients were biopsied in the older era, compared to 62/107 (58%) patients in the modern era.
Mean age at renal biopsy/diagnosis was 24 years for the older era, compared to 30 years in the later era. 51% of patients were male in the older era, compared to 60% of patients in the later era (Table 1). Other notable features in the table were that no patients were noted to have a lens defect in the earlier era, whereas 6/62 patients were noted to have a lens defect in the later era. Despite not reaching significance, median creatinine in the most recent era was 95 µmol/L compared 80 µmol/L in the older era.
Table 1: Comparison of patient characteristics according to the era of diagnosis. View Table 1
59% of patients reached ESKD during the period of study. The mean age at ESKD was 29 years for the older era versus 33 years for the later era (p = 0.21). Ten-year ESKD-free rate was 53.7% (95% C.I. 31.6-71.5%) for the earlier era, compared to 60.8% (95% C.I. 41.5-75.5%) for the later era (Figure 1).
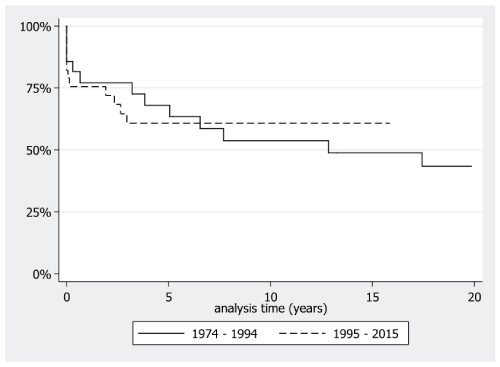 Figure 1: Kaplan-Meier curve of ESKD-free survival according to era of diagnosis. View Figure 1
Figure 1: Kaplan-Meier curve of ESKD-free survival according to era of diagnosis. View Figure 1
In multifactorial analysis, female sex was predictive of reduced ESKD risk as was lower serum creatinine at diagnosis (Table 2). Ten-year patient survival was 92.3% (95% C.I. 72.5-98.0%) in the first era compared to 97.1% (95% C.I. 80.9-99.6%) in the second (Figure 2). There was no effect of era on patient outcome and the only variable approaching significance was age at diagnosis (Table 3).
 Figure 2: Kaplan Meier curve of patient survival according to era of diagnosis. View Figure 2
Figure 2: Kaplan Meier curve of patient survival according to era of diagnosis. View Figure 2
Table 2: Multifactorial model of factors affecting time to ESKD for patients with Alport syndrome. View Table 2
Table 3: Multivariable model of factors affecting Alport patient survival. View Table 3
Though nearly a hundred years have passed since Alport syndrome was first described, it is only now that the heterogeneity of its disease spectrum is being fully understood. As outlined above, several genes responsible for collagen formation have been identified as causative; the type and number of mutations defines the severity of disease and its effect on men and women. Within our Irish national case series spanning 40 years of follow-up, genotyping was not widely performed; however we can see that many women were significantly affected including a number of patients who had not been known to have a family history of hereditary nephritis prior to diagnosis-hence a substantial number of patients demonstrated autosomal recessive features of this disease. Interestingly, relatively few women demonstrated hearing loss as part of their disease manifestations compared to men (13% women versus 50%).
59% patients progressed to end-stage kidney disease, however the majority of these were subsequently successfully transplanted with long transplant survival times. This indicates that early diagnosis and management can lead to favourable outcomes for this patient cohort. Temme, et al. [10] however found that patient survival in the Alport cohort in the ERA-EDTA registry was better than that of age-matched controls in their prospective study of dialysis patients from 14 national European registries between 1990-2009. The authors hypothesized that this was due to the absence of other vital organ involvement in Alport syndrome, as well as the lack of recurrence post-transplantation compared to other systemic inflammatory causes of end-stage kidney disease. There were several methodological differences between this study and our patient cohort, such as the exclusion of female cases from the final analysis. The much larger pan-European case numbers allowed the authors to perform much closer case-control matching, which may have allowed for a survival advantage for Alport patients to be shown more clearly, even when adjusted for age.
Mallett, et al. [15] performed a similar large cohort study of all ESKD patients with Alport syndrome in Australia and New Zealand, divided into an older (1965-1995) and more modern (1996-2010) era. Interestingly, they found that Alport syndrome was associated with superior dialysis patient survival to matched controls with other causes of ESKD during the period 1965-95. However, dialysis and renal transplant outcomes were comparable between RRT-treated Alport syndrome ESKD and matched non-Alport controls during the more recent period 1996-2010 due to relatively greater improvements in outcomes for non-Alport ESKD patients over time. This contrasts with our findings of similar patient survival in both eras, however this was not compared with non-Alport controls.
Our study of the clinical features and outcomes of Alport syndrome is one of the largest of its kind, following a national cohort of patients over a substantial 40-year time period. It was however limited by being retrospective in nature, requiring the participation of many nephrologists throughout Ireland to collect information that had not been recorded centrally in a contemporaneous manner. This may have led to recall bias, as well as selection bias due to loss of some patients to follow-up over time. Missing data were prevalent; in some cases we were able to impute these using last value brought forward, however for some variables this was not appropriate so columns were left blank. There was also a paucity of information available regarding treatment, including renin-angiotensin (RAAS) system blockade so we unfortunately were unable to analyse the effect this may have had on the results.
Alport syndrome is a rare inherited renal disease. In this large national cohort study spanning 40 years we have described the clinical features, renal-specific and overall patient outcomes for a large number of patients with Alport syndrome. A spectrum of disease activity is evident and a significant number of patients with likely autosomal recessive pattern of inheritance were included in the cohort. A large number of patients progressed to ESKD but the majority were transplanted with good long-term outcomes [16]. There was no difference in time to ESKD and in overall patient survival for Alport patients between the older and modern era. Indeed, overall patient survival with Alport syndrome is superior to that of patients with other forms of renal disease, though this is clearly influenced by the young age at disease presentation and diagnosis. This lends credence to the premise that early disease diagnosis and careful management can lead to favourable long-term outcomes for this patient group. In terms of future directions, we plan to genotype our national cohort to examine patterns of Alport disease inheritance in Ireland and to further assess the utility of genetic diagnosis for this disease. We would also like to examine the effect of therapeutic agents such as RAAS blockade on disease progression and we look forward to the potential introduction of other specific disease-modifying agents to ameliorate the progression of Alport syndrome in the future.
No funding was received for this research. The authors have no financial disclosures or other conflicts of interest to declare.
Research idea and study design-YK, AP, LW, PO'K, PC
Data acquisition-YK, AP, LW, SM, SK, MK, LC
Data analysis/interpretation-YK, BD, AD, PO'K, PC
Statistical analysis-YK, PO'K, PC
Supervision/mentorship-PC.
I declare that the results presented in this paper have not been published previously in whole or part, except in abstract format.
None of the authors of this paper have any conflicts of interest to declare.