Mutations in mitofusin 2 gene have been reported in Charcot-Marie-Tooth type 2 disease also known as Hereditary Sensory and Motor Neuropathy. With its cytogenetic location: 1p36.22.
A 43-year-old female with a family history of neuropathy was experiencing gradual deterioration and proximal weakness of the bilateral lower limb for the past 3 years. Her MRI scan (Brain and whole Spinal) was normal and Electromyography (EMG) report was suggestive of motor & sensory demyelinating polyneuropathy with features of segmental involvement. Her Creatine phosphokinase (CPK) levels were marginally elevated. However, all other investigations were within normal range.
A heterozygous missense variant denoted as c.701T > A at level of cDNA in exon 7 of the MFN2 gene was discovered which resulted in the substitution of Methionine by Lysine at position 234 [p.Met234Lys] of the amino acid sequence which was confirmed by Sanger sequence.
Hereditary motor and sensory neuropathies, Exome sequencing, MFN2 gene
Hereditary motor and sensory neuropathy with proximal dominance (HMSN-P) has been reported as a rare type of autosomal dominant adult-onset Charcot-Marie-Tooth disease. Hereditary motor sensory neuropathies (HMSN) are a group of heterogeneous diseases classified under motor neuropathies [1]. In CharcoteMarieeTooth disease (CMT) and the hereditary sensory neuropathies there is a significant sensory involvement along with distal lower motor neuron weakness [2]. The classical phenotype of HMSN is a length-dependent motor weakness and atrophy, primarily affecting the intrinsic foot muscles and the peroneal compartment of the leg, often leading to foot deformities such as pes cavus, pes planus, and clawing of the toes [3].
Mutations in the mitofusin 2 (MFN2) gene, which encodes a mitochondrial GTPase mitofusin protein, have recently been reported to cause both Charcot-Marie-Tooth 2A (CMT2A) and hereditary motor and sensory neuropathy VI (HMSN VI) [4].
A 43-year-old female with a family history of neuropathy presented with bilateral progressive proximal muscle weakness with sensory involvement. She first noticed reduced sensation and muscle weakness 3 years back and was clinically diagnosed to have hereditary sensory motor neuropathy. Her MRI scan (Brain and whole Spinal) was normal nerve conduction studies (NCS) and Electromyography (EMG) report was suggestive of motor & sensory demyelinating polyneuropathy with features of focal segmental involvement. Her creatinine phosphokinase (CPK) levels were elevated. However, all other investigations were within normal range. Probands father who had died at the age of 54 yrs. due to an unknown cause had symptoms of sensory motor neuropathy, which manifested at the age of 24 years. Probands two daughters age 14 and 17 years are asymptomatic and was found to be free of the mutation (Figure 1). Whole exome sequencing of the proband revealed a previously unreported heterozygous missense Variant in the MFN2 gene which is likely pathogenic.
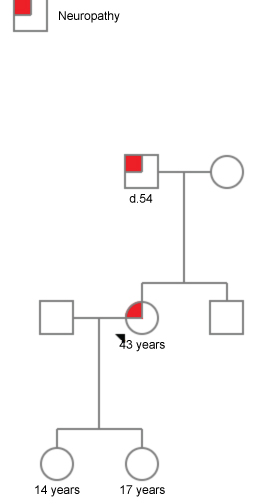 Figure 1: Pedigree of the probands' family with affected individuals.
View Figure 1
Figure 1: Pedigree of the probands' family with affected individuals.
View Figure 1
Whole exome sequencing (WES) of the proband's DNA was performed in line with the SureSelect® Human All Exon V6 kit on an Illumina® HiSeq 4000 Next Generation Sequencer. The DNA sequence was compared with the UCSC hg19 reference sequence. All reportable sequence variants were confirmed by visual inspection of the alignment.
The sequence reads were analyzed using in-house variant calling and annotation pipeline. Reads were aligned to a human reference sequence and processed using standard software. The data obtained were analyzed by annotation with multiple INFO fields required for filtering
(dbSNP allele frequencies (http://www.ncbi.nlm.nih.gov/projects/SNP/),
1000 Genomes allele frequencies (www.1000genomes.org),
Exome Aggregation Consortium (ExAC) allele frequencies (http://exac.broadinstitute.org),
the OMIM database and the Clinvar database (https://www.ncbi.nlm.nih.gov/clinvar/), using the SnpSift tool. Variant effect prediction was then performed using Genetic variant annotation and the effect prediction toolbox (SnpEff). Sanger sequencing was done to confirm the presence of the variants of interest by analyzing the chromatogram using Codon Code Aligner software.
In silico functional prediction was performed using Mutation Taster, SIFT, Polyphen2, and Provean.
A heterozygous missense mutation denoted as c.701T > A at level of cDNA in exon 7 of the MFN2 gene was discovered which resulted in the substitution of Methionine by Lysine at position 234 [p.Met234Lys] of the amino acid sequence was confirmed by Sanger sequence (Figure 2). Substitution of methionine to Lysine changes the physio-chemical characteristics of the amino acid, thus causes a non-conservative amino acid substitution.
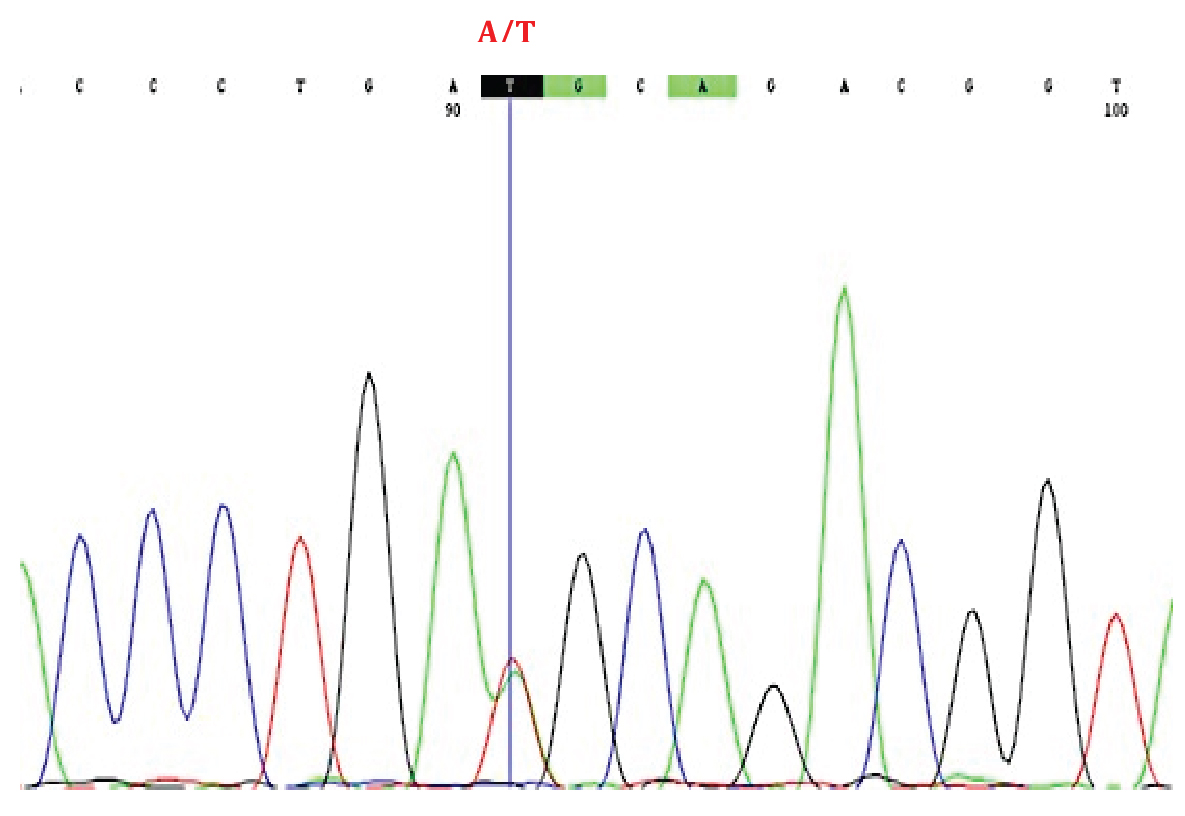 Figure 2: Sanger sequence chromatogram showing a novel homozygous missense mutation in exon 7 of the MFN2 gene, [MIM: 609260]: c.587C > T [NP_055689.1: p.Met234Lys] associated with hereditary motor and sensory neuropathy.
View Figure 2
Figure 2: Sanger sequence chromatogram showing a novel homozygous missense mutation in exon 7 of the MFN2 gene, [MIM: 609260]: c.587C > T [NP_055689.1: p.Met234Lys] associated with hereditary motor and sensory neuropathy.
View Figure 2
This substitution in a highly conserved region of the MFN2 gene (phastCons100way_vertebrate score = 1; PhyloP score = 5.145) and majority of in-silico functional prediction tools concluded this variant as pathogenic (Table 1).
Table 1: Results of in silico mutation prediction analysis. View Table 1
Charcot-Marie-Tooth disease (CMT), also named hereditary motor and sensory neuropathies, includes a clinically and genetically heterogeneous group of disorders affecting the peripheral nervous system, with a relatively high prevalence of 1:2,500 [5]. The main forms of CMT have been subdivided into primary demyelinating neuropathies (myelinopathies), which include CMT1 (autosomal-dominant inheritance), CMT1X (X-linked), and CMT4 (autosomal-recessive), and neuropathies characterized by primary axonal defects such as CMT2 (autosomal-dominant or recessive). Additional sub-forms of hypomyelinating and/or dysmyelinating neuropathies include Dejerine-Sottas syndrome (DSS, also called CMT3), hereditary neuropathies with liability to pressure palsies (HNPP), and congenital hypomyelination (CH) [6]. Mutations in mitofusin 2 (MFN2) have been reported in Charcot-Marie-Tooth type 2 (CMT2) families. In patients with a documented family history of CMT2 the frequency of MFN2 mutations was 33% indicating that MFN2 mutations are a major cause in this population [7].
As with our patient most suffer from demyelinating forms of CMT.
However, investigations of the underlying molecular and cellular disease mechanisms, employing cell cultures and animal models have revealed an overlap of pathological signs between myelinopathies and axonopathies [8] Charcot-Marie-Tooth disease type 2A was previously mapped to an interval containing KIF1B. New studies have shown that CMT2A patients contain a loss-of-function mutation in the motor domain of the KIF1B gene. This is clear indication that defects in axonal transport due to a mutated motor protein can underlie human peripheral neuropathy [9]. However, the gene responsible for CMT2A is still controversial, and the mechanism of pathogenesis remains uncertain. It has also been shown that the receptor tyrosine kinase IGF1R is a new direct binding partner of KIF1Bβ, and its binding and transport is specially impaired by the Y1087C mutation of KIF1Bβ, which we detected in hereditary neuropathic patients [10]. Approximately ten genes have now been identified to cause CMT disease [11]. Whole exome sequencing (WES) of the proband revealed a novel variant in the MFN2 gene, c.701T > A (p.Met234Lys).
MFN2 is a mitochondrial transmembrane GTPase, which regulates mitochondrial architecture by fusion of mitochondria and most pathological variants of this gene is located within or immediately upstream of the GTPase domain or within two coiled-coil domains, which are critical for the functioning or mitochondrial targeting of MFN2. Formation of a mitochondrial network is essential for a proper functioning PNS [11]. Even though there is no current treatment for the condition recent studies have shown that antisense oligonucleotides (ASOs) offer new hope for patients with Charcot-Marie-Tooth disease type 1A (CMT1A) [12]. In conclusion we report a novel heterozygous missense variant in exon 7 of the MFN2 gene causing CMT disease.
Ethics approval for the study was obtained by the Faculty of Medicine, University of Colombo Ethics Review Committee (EC-16-179). Duly filled consent forms are available with the corresponding author.
Written informed consent to publish this material was obtained from the proband and her two daughters.
Data is not available in a public domain it will be shared on request from interested parties.
Authors declare that there are no competing interests.
There was no funding for this study.
DH and VHWD were the clinicians looking after the patient. BAPSP and KW performed laboratory testing. NN performed bioinformatics analysis. DH wrote the first draft of the manuscript with contributions from all. All authors reviewed, modified and approved the final version of the manuscript.
We wish to acknowledge the referring doctor and the laboratory staff for their contribution.