RAD51a is a highly conserved protein and its major role is the repair of DNA double strand breaks. Endogenous species are generated during normal cell metabolic activities and can cause damage to DNA, as well as several environmental factors. The interactions RAD51a perform with other proteins help the maintenance of oncogenetic metabolism within cells. RAD51a interacts with PCNA, FANCD2 and ABL1, among many other cancer-related proteins. PCNA acts as a DNA clamp and is related to the replication process, FANCD2 arrests DNA replication fork progression in response to DNA damage and ABL1 is a proto-oncogene related to cell differentiation. Protein-protein interactions (PPIs) are governed by the presence of hot spots within the interface of interaction. Identifying residues directly involved in PPIs enables the likelihood of modulating such complexes with biologically active small molecules such as synthetic peptides, which leads to a new era of diseases treatment. Here, we use an in silico approach to determine the best free-energy of interaction between RAD51a and the targeted cancer-related proteins PCNA, FANCD2 and two chains of ABL1. We propose an interaction interface between RAD51a and those proteins and identified hot spots that could be useful to understand the molecular basis of their interaction. We believe that further studies may find small-targeted molecules with therapeutics properties that could modulate those interactions and increase our knowledge regarding the complex trait diseases such as cancer.
RAD51a, PCNA, FANCD2, ABL1, Protein-protein interactions
RAD51a is a highly conserved protein and its major role is the repair of DNA double strand breaks (DSBs) [1]. Genome integrity is maintained by a series of metabolic events in which cells recognize and amend DNA molecules that is damaged due to either endogenous or exogenous agents [2,3]. Endogenous reactive oxygen species (ROS) are generated during normal cell metabolic activities [4] and exogenous damage can be caused by radiation [5,6], chemicals [7,8] and other environmental factors [9,10]. DNA damage leads to a structural impairment of the double helix molecule [11] and consequently affects the ability of the cell to translate the information encoded within genetic material. In addition, DNA damage can lead to mutations in the genome and alter the survivability rate of daughter cells during cell division [12].
RAD51a takes part in DNA repair through homologous recombination. The protein contains 339 amino acids and folds into a heptameric structure formed by identical polypeptide chains (Figure 1A). The 3-D (three-dimensional) structure of RAD51a is related to its DNA repair function. When a DNA DSB takes place, RAD51a searches for homology and catalyzes strand pairing in order to repair the damage. The RAD51a structure establishes a helical nucleoprotein filament around the DNA [13] can bind to other proteins to accomplish its function [14,15]. RAD51a interacts with proteins related to the DNA metabolism, such as the FA Complementation Group D (FANCD2), the proliferating cell nuclear antigen (PCNA) [16,17] and the Abelson murine leukemia viral oncogene homolog 1 (ABL1) [18-20]. All of them are somehow related to the metabolism of DNA and anomalies within their sequence or disruption of the interactions they perform with other proteins may increase cancer and other diseases susceptibility.
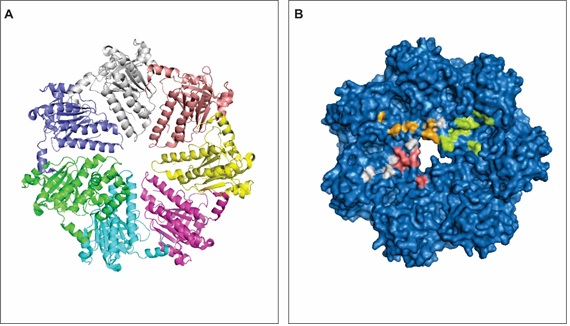 Figure 1: RAD51a 3-D structure. A - The heptameric structure of RAD51a. Each color represents an identical polypeptide chain of the protein. B - 3-D structure showing the surface of RAD1a and the predicted hot spots for its interaction with the PCNA (orange), FANCD2 (white), SH3 domain (pink) and tyrosine kinase domain (green) of ABL1. View Figure 1
Figure 1: RAD51a 3-D structure. A - The heptameric structure of RAD51a. Each color represents an identical polypeptide chain of the protein. B - 3-D structure showing the surface of RAD1a and the predicted hot spots for its interaction with the PCNA (orange), FANCD2 (white), SH3 domain (pink) and tyrosine kinase domain (green) of ABL1. View Figure 1
Cancer is a prevalent disease worldwide. Over 90.5 million people had cancer in 2015 [21]. It is estimated that over 14.1 million new cases of cancer occur every year, leading to 8.8 million deaths [22]. RAD51a, PCNA, FANCD2 and ABL1 are potential therapeutic targets in cancer therapy [23-26] as they are related to several types of cancer. PCNA acts as a DNA clamp and is related to the replication process, FANCD2 arrests DNA replication fork progression in response to DNA damage and ABL1 is a proto-oncogene related to cell differentiation. These proteins interact with a variety of molecules in order to maintain genomic stability. Protein-protein interactions (PPIs) are a key factor for a proper DNA metabolism and they regulate a wide range of biochemical pathways and processes, including homologous recombination [27], cell division, cell cycle progression [28] and cancer onset [29].
PPIs help to understand how proteins contribute to dysregulated oncogenic pathways and disease progression. Moreover, several PPIs related to nuclear metabolism show therapeutic relevance, mainly regarding cancer, and have been targeted by high-throughput approaches in order to generate new insights on the increasing biological data [30]. Experimental and in silico approaches related to protein networks have contributed to the genetic and molecular classification of the most prevalent cancers in humans [31]. In addition, cancer-related proteins are therapeutically targeted through the inhibition of their molecular interactions. Therefore, the study of disease-specific protein interaction network is vital to direct cancer treatment [31,32].
Here, we use an in silico approach to determine the best free-energy of interaction between RAD51a and the targeted cancer-related proteins PCNA, FANCD2 and two chains of ABL1. We propose an interaction interface between RAD51a and those proteins and identified hots pots that could be useful to understand the molecular basis of their interaction.
All the 3-D structures used in the analysis are available in the PDB (protein databank; https://www.rcsb.org/). We used KBDOCK in order to find protein domains and possible interaction between protein domains [33]. The protein docking was performed by ClusPro [34]. We used PyMol (https://pymol.org) for the visualization of the interface of interaction and the visualization of hot spots and polymorphic residues. The hot spots in the proteins under study were identified by KFC2. The server offers an automated analysis of a protein complex interface. The server analyses the structural environment around amino acid residues and checks for already known hot spots environments determined experimentally. The hot spot prediction is based on characteristics regarding conformation specificity (K-FADE) and biochemical features such as hydrophobicity (K-CON) [35,36]. Finally, the polymorphic residues were identified through the dbSNP (data base of single nucleotide polymorphism; https://www.ncbi.nlm.nih.gov/SNP).
PPIs are governed by hot spots that are present on the interface of interaction. Hot spots are amino acid residues that account to the binding free energy of a certain protein [35,36] and is the site that has a higher probability for ligands to interact [37,38]. Analyzing the interaction interface between RAD51a and its binding partners, we identified several clusters of hot spots (Table 1 and Figure 1B). Identifying residues directly involved in PPIs enables the likelihood of modulating such complexes with biologically active small molecules such as synthetic peptides [39]. Moreover, the development of such techniques is promising for the therapeutics of complex diseases like cancer [40-44], Alzheimer's disease [45], Parkinson's disease [46], epilepsy [47] and infection [48].
Table 1: Cluster of hot spot residues on RAD51a interface of interaction. View Table 1
Hot spots govern thermodynamic processes related to PPIs. The amino acid residues identified as hot spots on the RAD51a interaction interfaces define the biochemical properties involved in its interactions with PCNA, FANCFD2 and ALB1 (Figure 1B). Solvation potential, residue interface propensity, hydrophobicity, planarity, protrusion and accessible surface area regulate those interactions and guarantee or not the functions those proteins exert in the complex.
PCNA is highly conserved among species and required for a proper DNA replication [49]. This protein surrounds the DNA and recruit partner proteins related to DNA metabolism such as replicatio [50], repair [50,51], chromatin remodeling [52] and epigenetics [53]. PCNA has 261 amino acid residues and 3 identical units of a polypeptide (Figure 2A), organized in a way that facilitates it to encircle the DNA when PCNA is active.
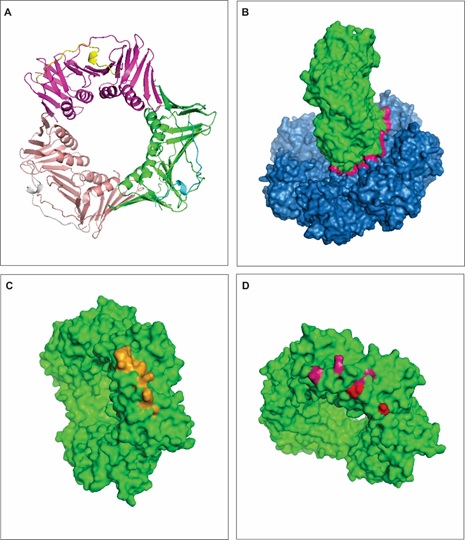 Figure 2: Interaction between RAD51a and PCNA. A - 3-D structure of PCNA. PCNA is a protein trimer, each chain is representing by a different color. B - The interface of interaction between RAD51a (blue) and PCNA (green). The interface of interaction in the contact region between the interacting proteins (pink), which is characterized by a large region without empty spaces or pockets. C - The figure represents in orange the predicted hot spots on the PCNA protein (green) when it interacts with RAD51a. D - The figure shows highly polymorphic residues that overlaps hot spots (pink) or are present in the proximity of hot spots (red). View Figure 2
Figure 2: Interaction between RAD51a and PCNA. A - 3-D structure of PCNA. PCNA is a protein trimer, each chain is representing by a different color. B - The interface of interaction between RAD51a (blue) and PCNA (green). The interface of interaction in the contact region between the interacting proteins (pink), which is characterized by a large region without empty spaces or pockets. C - The figure represents in orange the predicted hot spots on the PCNA protein (green) when it interacts with RAD51a. D - The figure shows highly polymorphic residues that overlaps hot spots (pink) or are present in the proximity of hot spots (red). View Figure 2
The interfaces of PPIs are normally large and with no empty spaces or pockets (Figure 2B) compared to binding sites for small molecules [54,55]. Conserved amino acid residues, the so-called hot spots, stabilize the RAD51a-PCNA complex (Table 2; Figure 2C) and this interaction guarantees a high-fidelity repair of DSBs on the DNA. DNA repair is essential to recover genetic information that would be possibly lost during natural processes that induce DBSs, such as replication and homologous recombination. Figure 2B shows a model for the interaction between RAD51a and PCNA. It has been suggested that the interaction between these two proteins are regulatory, and RAD51a inhibits PCNA in order to arrest the replication process [56-58].
Table 2: Cluster of hot spot residues on PCNA interface of interaction. View Table 2
Disruption of the thermodynamic balance within the RAD51a-PCNA complex may be a cause for disease onset. Mutations and polymorphisms in either proteins could affect the free-energy pattern maintained by hot spots in the interaction interface and induce cancer development. More drastic consequences would be seen if the conserved amino acid residues (Table 3) are affected. Figure 2D shows some commons single nucleotide polymorphisms (SNPs) that take place on PCNA residues. We identified 7 hot spot residues on the PCNA interface of interaction with RAD51a and among them, TYR 151, LYS 154, LEU 157 are highly polymorphic. Figure 2D also shows a couple of polymorphic residues that are not classified as hot spots but are in the proximity and may affect the free energy of PCNA binding to RAD51a. SNPS in such residues may regulate PNCA interaction with other proteins and its expression. PNCA polymorphisms have been found in patients with several types of cancer [59,60].
Table 3: Cluster of hot spot residues on FANCD2 interface of interaction. View Table 3
The protein FANCD2 is ubiquitinated as a response to DNA damage, which is fundamental for the DNA repair. FANCD2 activity drives the homology-directed DNA repair process [61]. The result of its expression is arrest of DNA replication fork progression, protects cells from chromosome abnormalities and loss of genetic information during cell division and gamete formation [62]. The protein folds into a 3-D structure with more than 50% of helical chains (Figure 3A). The FANC protein family include several representatives with several isoforms and low levels of similarity due to the diversity of functions they present [63].
 Figure 3: Interaction between RAD51a and FANCD2. A - 3-D structure of FANCD2. B - The interface of interaction (pink) between RAD51a (blue) and FANCD2 (orange). C - The figure represents in pink the predicted hot spots on the FANCD2 protein (orange) when it interacts with RAD51a. D - The figure shows highly polymorphic residues that overlaps hot spots (pink) or are present in the proximity of hot spots (red). View Figure 3
Figure 3: Interaction between RAD51a and FANCD2. A - 3-D structure of FANCD2. B - The interface of interaction (pink) between RAD51a (blue) and FANCD2 (orange). C - The figure represents in pink the predicted hot spots on the FANCD2 protein (orange) when it interacts with RAD51a. D - The figure shows highly polymorphic residues that overlaps hot spots (pink) or are present in the proximity of hot spots (red). View Figure 3
Figure 3B shows the interface of interaction between RAD51a and FANCD2. FANCD2 is known to stabilize the complex formed by RAD51 and DNA. The protein binds to RAD51 in a way that it protects the 5'-DNA end [17]. Patients with a disrupted structure of FANCD2, mainly carriers of Fanconi anemia, have higher risk of cancer development [64,65]. The most frequent cancer types related to FANCD2 are ovarian [66], hepatic [67], lung [68], breast cancer [69] and leukemia [70]. FANCD2 co-localizes with RAD51a following DNA damage is an indication that FANCD2 and RAD51a share roles in DNA repair through physical interactions [16,17,71].
We found 9 conserved hot spot residues (Figure 3C) within the interaction interface of FANCD2 and RAD51a (Table 3). Among those, 5 have been identified as polymorphic residues as HIS 594 can be changed into Gly, ILE 855 into Val, ARG 879 into Lys, ARG 886 into Pro and finally ILE 849 which corresponds to a synonymous mutation (Figure 3D). Polymorphisms and mutations in FANCD2 genes have been reported to be related to cancer and other diseases [72-75].
The ABL1 protein has two main domains, a tyrosine kinase domain and a SH3 domain. The former has regulatory functions to other proteins [76], is related to division, proliferation, apoptosis and differentiation [77] while the latter regulates ABL1 expression and protein-protein interactions [78]. The SH3 domain folds into a highly conserved 3-D structure (Figure 4A) arranged in β-strands and anti-parallel β sheets chains [79]. The SH3 domain contains conserved aromatic amino acid that build up a hydrophobic center and an aliphatic pocket on the surface of the protein. These features are important for a correct folding of the protein and for the free-energy binding patter of ABL1 [80]. ABL1 is characterized as proto-oncogene and disruption of SH3 domain turns the protein into an oncogene related to onset of several type of cancers [25]. The tyrosine kinase domain of ABL1 is also highly conserved across species [81] and a large variety of kinase inhibitors have been proposed for the treatment of genetic-related anomalies [82]. Some common patterns in the protein kinase domain are recognized in several proteins, such as an ATP binding region, conserved residues, a catalytic site and peptide sequences [83]. The 3-D structure for the ABL1 tyrosine kinase is represented in Figure 4B.
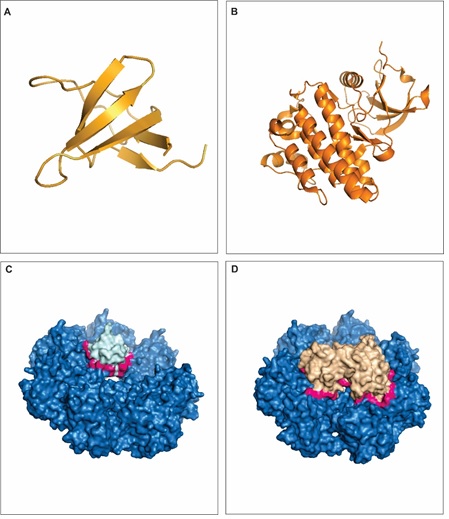 Figure 4: Interaction between RAD51a and ABL1. A - 3-D structure of ABL1 SH3 domain. B - 3-D structure of ABL1 tyrosine kinase domain. C - The interface of interaction (pink) between RAD51a (dark blue) and ABL1 SH3 domain (light blue). D - The interface of interaction (pink) between RAD51a (dark) and ABL1 SH3 domain (light brown). View Figure 4
Figure 4: Interaction between RAD51a and ABL1. A - 3-D structure of ABL1 SH3 domain. B - 3-D structure of ABL1 tyrosine kinase domain. C - The interface of interaction (pink) between RAD51a (dark blue) and ABL1 SH3 domain (light blue). D - The interface of interaction (pink) between RAD51a (dark) and ABL1 SH3 domain (light brown). View Figure 4
Both the SH3 and the tyrosine kinase domains of the protein ABL1 interacts with RAD51a and the interactions are related to DNA repair [20,84]. Interestingly, RAD51a also contains the SH3 and the tyrosine kinase domains. The interface of interaction between RAD51a and ABL1 SH3 domain is represented in Figure 4C and the interface of interaction between RAD51a and ABL1 tyrosine kinase domain is represented in Figure 4D. ABL1 interaction with RAD51a guarantees a faithful DSBs repair and genomic stability, in a way that it helps to achieve a proper functioning of oncogene metabolism [20,85]. Moreover, it has been shown that RAD51a is regulated by tyrosine phosphorylation at residues 54 and 315 [86]. Here, we found a hot spot on Tyr 314 (Table 1), which may as well contribute to the interaction of ABL1 and RAD51a.
We found 4 conserved hot spot residues (Figure 5A) within the interaction interface of ABL1 SH3 and RAD51a and 10 conserved hot spot residues (Figure 5B) within the interaction interface of ABL1 tyrosine kinase and RAD51a (Table 4). Among those, 5 have been identified as polymorphic residues as Met 437 can be changed into Ile, Ile 443 into Thr, Tyr 456 into Cys, Glu 459 into Lys and Leu 451 as a synonymous mutation (Figure 5C).
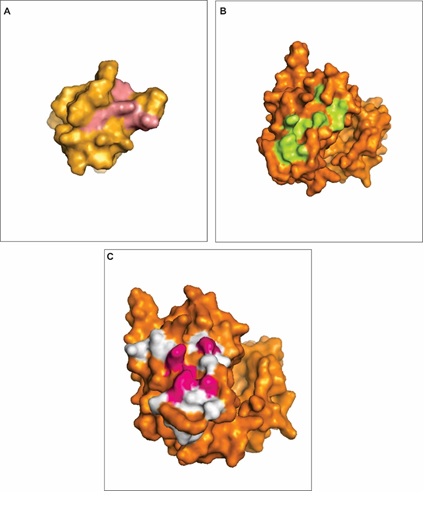 Figure 5: Hot spots prediction in ABL1 domains. A - The figure represents in pink the predicted hot spots on the ABL1 SH3 domain (orange) when it interacts with RAD51a. B - The figure represents in pink the predicted hot spots on the ABL1 tyrosine kinase domain (orange) when it interacts with RAD51a. C - The figure shows highly polymorphic residues that overlaps hot spots (pink) or are present in the proximity of hot spots (white) in the ABL1 tyrosine kinase domain (orange). View Figure 5
Figure 5: Hot spots prediction in ABL1 domains. A - The figure represents in pink the predicted hot spots on the ABL1 SH3 domain (orange) when it interacts with RAD51a. B - The figure represents in pink the predicted hot spots on the ABL1 tyrosine kinase domain (orange) when it interacts with RAD51a. C - The figure shows highly polymorphic residues that overlaps hot spots (pink) or are present in the proximity of hot spots (white) in the ABL1 tyrosine kinase domain (orange). View Figure 5
Table 4: Cluster of hot spot residues on ABL1 interface of interaction. View Table 4
Cancer is a prevalent disease worldwide. RAD51a, PCNA, FANCD2 and ABL1 are potential therapeutic targets in cancer therapy. These proteins interact with a variety of molecules in order to maintain genomic stability. Protein-protein interactions (PPIs) are a key factor for a proper DNA metabolism and they regulate a wide range of biochemical pathways and processes. Here, we have shown the in silico interaction between RAD51a and the cancer-related proteins PCNA, FANCD2 and ABL1 and we presented the most favorable free binding energy of hot spots important for those interactions. We believe that further studies may find small-targeted molecules with therapeutics properties that could modulate those interactions and increase our knowledge regarding the complex trait diseases such as cancer.