Mixed Connective Tissue Disease is an autoimmune clinical entity with characteristics of overlap syndrome including: Systemic Lupus Erythematosus, Rheumatoid Arthritis, Systemic Sclerosis, Polymyositis and Dermatomyositis. There is an active controversy as to whether it is a variant of Lupus or it is a different entity. The objective of these case reviews is to describe clinical-serological characteristics of 6 patients from AGAR clinic (Guatemalan Association against Rheumatic Diseases) that meet the criteria for this syndrome according to Alarcón-Segovia and Villareal criteria. Although the controversy persists, after an extensive review of the literature we consider that Mixed Connective Tissue Disease is not a variant of lupus, but a different entity with a different course and prognosis.
Alarcón-Segovia and Villareal, Anti-RNP, Mixed connective tissue disease, Gordon sharp
ACEI: Angiotensin Converting Enzyme Inhibitors; ACPA: Anti-Cyclic Citrullinated Peptide Antibody; AGAR: Guatemalan Association against Rheumatic Diseases; Anti-RNP: Ribonucleoprotein Antibody; ARA II: Angiotensin II Receptor Antagonist; CK: Creatine Kinase; CRP: C-Reactive Protein; DM: Dermatomyositis; DMARDS: Disease Modifying Antirheumatic Drugs; ENA: Extractable Nuclear Antigen; ESR: Erythrosedimentation Rate; FANA: Fluorescence Anti-Nuclear Antibody; HLA: Human Leukocyte Antigen; ILD: Interstitial Lung Disease; LDH: Lactate Dehydrogenase; MCTD: Mixed Connective Tissue Disease; NSAIDs: Nonsteroidal Anti-Inflammatory Drugs; PAH: Pulmonary Arterial Hypertension; PM: Polymyositis; RA: Rheumatoid Arthritis; RF: Rheumatoid Factor; RP: Raynaud's Phenomenon; SLE: Systemic Lupus Erythematosus; SS: Sjögren's Syndrome; SSc: Systemic Sclerosis; U1sn-RNP: Uridyl Rich Small Nuclear Ribonucleoprotein
Mixed Connective Tissue Disease (MCTD) was first described in 1972 by Gordon Sharp, et al., conducting a study in 25 patients who had clinical characteristics of several rheumatic diseases: Systemic Lupus Erythematosus (SLE), Systemic sclerosis (SSc) and Polymyositis (PM) [1,2]. Extractable Nuclear Antigen (ENA) now called Ribonucleoproteins (RNP) are a group of cytoplasmic and nuclear antigens of whichAnti-U1 RNP (Ribonucleoprotein) is the characteristic of MCTD. The common feature of these patients is that they possessed Anti-U1 RNP in very high titres above 1:1000. All being Anti-Sm negative, and unlike the SLE patients where Anti-RNP positive but with lower values [2,3]. Another finding was that there was no renal involvement. Antinuclear antibodies (FANA) had speckled pattern predominance unlike patients with SLE [1]. The epidemiology shows a clear female predominance 5:1 [4]. The incidence of cases varies in each region from 0.2-0.8 cases per 100,000 people [5-7].
In patients with U1-SnRNP positive it has been found a discrete genetic association with HLA-DR4/Gm (1,3; 5,21) [8], also an increase in anti-U1-snRNP autoantibodies of IgG type [3]. In a Mexican population, an association of HLA-A2 and HLA-B35 was found to be the most frequent MHC class I alleles in patients with MCTD, although they were not statistically significant, the most frequent DQ allele in the patients were DQ1, suggesting a particular genetic association for MCTD [6,9].
Nucleosomes are compact building blocks of chromatin and consist of an octamer of two copies of histones H2A, H2B, H3 and H4, wrapped around approximately 146 base pairs of DNA [10]. U1-RNP is the target of autoantibodies that meet the classification criteria for anti-RNP antibodies in MCTD. This complex of nuclear macromolecules plays an essential role in splicing pre-mRNA in mRNA. U1-RNP is composed of an RNA skeleton, U1-RNA, three proteins that are highly specific for U1-RNP (proteins U1-A, U1-C and U1-70kD), plus a series of additional proteins. This is common to multiple macromolecules that splice U-RNP and RNA (Figure 1) [11].
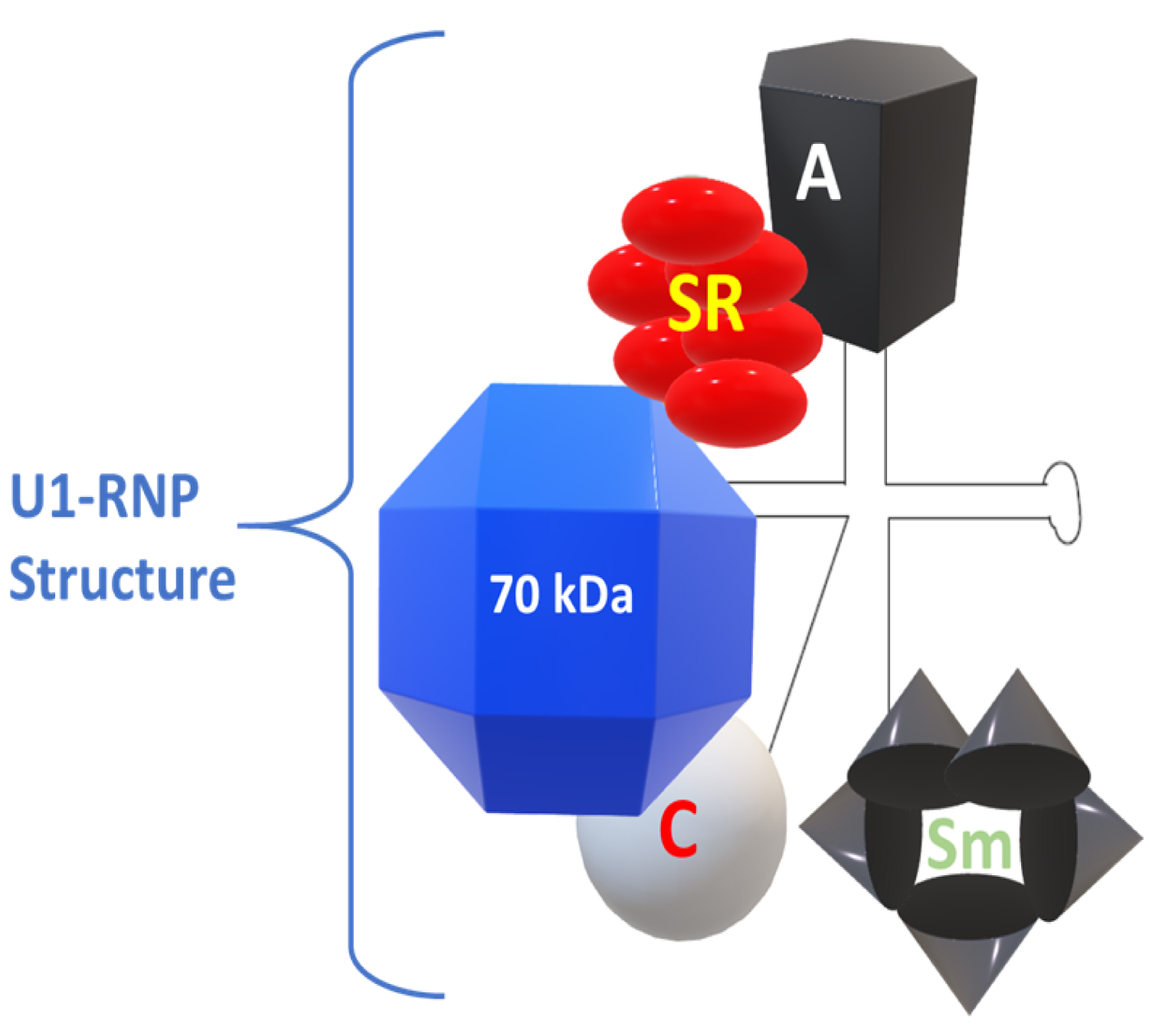 Figure 1: The structure of U1-RNP. Modified from Greidinger EL, Hoffman RW [11].
View Figure 1
Figure 1: The structure of U1-RNP. Modified from Greidinger EL, Hoffman RW [11].
View Figure 1
Spliceosome is a large complex consisting of proteins and RNA that catalyzes the removal of introns from pre-messenger RNA (preRNA) and splicing exons coding to produce mature mRNA. U1, U2, U5 and U4/U6 are small nuclear ribonucleoproteic particles (snRNP). These particles are assembled to the preRNA substrate together with another type of protein to form the spliceosome [12,13]. Autoimmunity to specific components of the spliceosome is the immunological characteristic of MCTD.
The snRNPs contain small RNA species that vary in size from 80 to 350 nucleotides that are composed of proteins. These RNAs contain a high uridine content and, therefore, are called U-RNA; five different U-RNAs were defined with the basis of immunoprecipitation (U1, U2, U4, U5 and U6). Autoantibodies to these complexes primarily target the protein components. Anti-Sm antibodies precipitate five proteins with molecular weights of 28,000 (B'B), 16,000 (D), 13,000 (E), 12,000 (F) and 11,000 (G); five of these polypeptides are common to RNAs U1, U2, U4, U5 and U6. Anti-RNP antibodies precipitate three proteins with molecular weights of 68,000 (70K), 33,000 (A') and 22,000 (C); these polypeptides are associated only with U1 RNA. The clinical features considered distinctive of MCTD are associated with the presence of antibodies with specificity of 70 kD, with an immunodominant epitope that encompasses amino acid residue 125 flanked by important conformational residues at positions 119 to 126 [14].
Epitope spreading is the process that occurs when a patient who previously had positivity to a given antibody recognizes another antibody as positive during follow-up; if the new autoantibody recognizes an epitope related to another epitope for which the patient previously had autoantibodies [12,15]. This phenomenon can occur intramolecularly, when the propagation of reactivity occurs within a single autoantigen with multiple epitopes in the same protein, or intermolecularly, when the reactivity extends to other polypeptides or proteins within a macromolecular complex. This phenomenon is highly prevalent in patients with MCTD [15-17] (Figure 2).
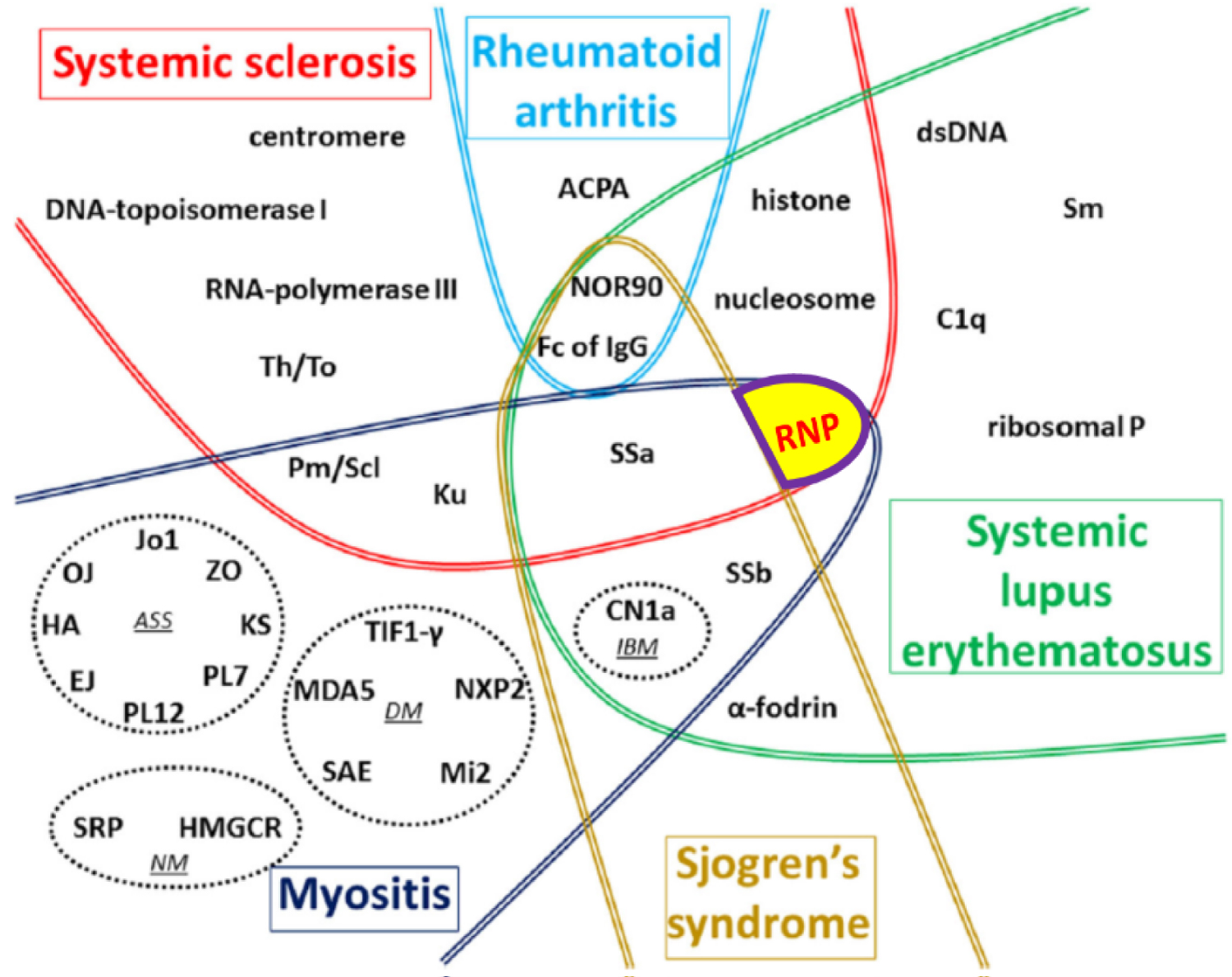 Figure 2: Global vision of autoantibodies target autoantibodies (AAb) according to the type of connective tissue diseases (CTD). Adapted with permission from Didier K, Bolko L, Giusti D, Toquet S, Robbins A, Antonicelli F (2018) Autoantibodies Associated with Connective Tissue Diseases [16].
View Figure 2
Figure 2: Global vision of autoantibodies target autoantibodies (AAb) according to the type of connective tissue diseases (CTD). Adapted with permission from Didier K, Bolko L, Giusti D, Toquet S, Robbins A, Antonicelli F (2018) Autoantibodies Associated with Connective Tissue Diseases [16].
View Figure 2
The diagnosis of MCTD is often a challenge for clinicians, due to the overlap and the number of initial symptoms, with variable courses [18,19]. For this reason, several diagnostic criteria have been proposed, one of the most accepted is Alarcón-Segovia, with a sensitivity and specificity of 62.5% and 86.2%, respectively [20,21] and in this case, we have selected them for their simplicity and easy application.
Several authors do not consider the definition of MCTD a distinctive clinical entity, including Reichlin, et al. [22], LeRoy, et al. [23] and Nimelstein, et al. [24], since some the clinical characteristics of MCTD do not differ from those of SLE patients with positive anti-snRNP antibodies and many MCTD patients progress to SLE or SSc during their evolution [25-27]. However, in Susana Capelli's study [28]; One hundred and sixty-one patients with MCTD were retrospectively evaluated in diagnosis and evolution, they were followed over 7.9 years, concluding that more than half of the patients remained with MCTD [28].
At AGAR clinic (Guatemalan Association against Rheumatic Diseases) in Guatemala City that specializes in the care of rheumatic conditions; we reviewed our cases of MCTD.
The objective of this study is to describe the clinical-serological characteristics of patients with MCTD that met criteria of Alarcón-Segovia and Villareal [20].
Case based review registry of 6 patients with MCTD. A review of the cases of the AGAR clinic; 6 cases met criteria for MCTD, over a 10-year period, from 2009 to 2019.
The patients included in this review were required to meet the criteria of Alarcón-Segovia listed below:
1. Serological criteria: Positive antibodies to U1 RNP antibodies in a titer ≥ 1:1600 dilution
2. Clinical criteria:
a) Swelling of the hands (puffy hands)
b) Synovitis
c) Myositis
d) Raynaud's phenomenon
e) Sclerodactyly with or without proximal scleroderma
Serological criteria and ≥ 3 of the clinical criteria (coexisting edema of the hands, Raynaud's phenomenon and sclerodactyly require additional compliance with criteria 2 or 3) [20].
Exclusion criteria:
1. Patients who do not fulfill clinical criteria for MCTD.
2. No confirmatory serological tests.
3. No appropriate follow-up.
The patients studied correspond to patients from various areas of the country, of which 50% came from the urban area and the other 50% from the rural area. The ages at the time were in a range between 25 and 71 years old with a mean of 45, the majority being mestizo population and only 1 case of indigenous origin. All the patients studied are female. The time of evolution of clinical features varies from 1 to 10 years since the appearance of the first clinical manifestations; being the time of 1 to 5 years for three cases and the rest of 5 to 10 years of evolution.
Regarding the clinical manifestations for diagnosis of MCTD, using the criteria of Alarcón Segovia and Villareal; It was found that 5 of the 6 patients presented the defining criteria: Hand swelling, synovitis and Raynaud. Only one of the cases presented myositis symptoms. Half of the cases presented sclerodactyly, however; these cases were associated with Raynaud's phenomenon as well. All cases presented positivity for Anti RNP whose titers exceed 1:1600 dilution (Table 1).
Table 1: Alarcón-Segovia and Villareal criteria applied to our clinical cases. View Table 1
Several clinical manifestations were presented that were not limited solely to the criteria of Alarcón-Segovia and Villareal. Therefore, a broader clinical feature stands out from initial manifestations to the present, with signs and symptoms referred to different biophysical spheres or domains. It is observed in all cases the presence of fever either referred or verified in the clinical history, as well as asthenia in all cases. Signs/symptoms such as: Alopecia, xerophthalmia, weight loss, skin rash and anasarca occurred in 5 of the 6 patients. Symptoms such as anorexia, xerostomia, headache, dyspnea, and photosensitivity in 3 of the 6 cases.
We observed that all the patients had at some time FANA positive 1:80, with a speckled pattern. Anti-DNA antibody positivity was found in three patients, none were positive for Anti-Sm. Rheumatoid Factor (FR) was positive in all patients studied, in high titres; however, none were positive for Anti-cyclic citrullinated peptide Antibody (ACPA). Acute phase reactants were found elevated in only two patients. No patients had hypocomplementemia. There was no positivity for Anti-ScL70 or Anti-Jo. Total creatine kinase was raised only in one case, which was the one that debuted with myositis, although there was no elevation of Aldolase or LDH. Anti-RNP was positive in all cases with titles greater than 1:1600. Two patients with positivity for Anti-SSA/Ro were found, however, there was no positivity for Anti-SSB/La (Table 2).
Table 2: Serological studies performed and most common laboratory findings. View Table 2
Common laboratory alterations found were hematology and urinalysis in 4 patients, 2 had normal routine clinical laboratory studies. The findings correspond to 3 patients with leukopenia. Clinical case number 1 presented: Anemia, lymphopenia and significant moderate proteinuria. Case number 2 presented thrombocytopenia and macroscopic hematuria; and case number 6 presented neutropenia as the only initial laboratory finding. Clinical case 3 and 4 did not show initial laboratory alterations (Table 2).
Treatment consisted of a combination of a DMARDS, NSAIDs, steroids in some cases: Calcium-antagonists, ARA II, ACEI, phosphodiesterase 5 inhibitors, prokinetic and proton pump inhibitors.
DMARDS were used for arthritis control when there was a predominant clinical SLE like features. Mycophenolate mofetil was used in case 2 because it was the most complicated case due to cardiopulmonary involvement, with the need to use in addition phosphodiesterase 5 inhibitor. Drugs were used for specific clinical situations such as Raynaud's phenomenon, with use of calcium antagonists. In addition, proton pump inhibitors for control of gastroesophageal reflux.
Case 2 presents radiological findings, later confirmed with high-resolution computed axial tomography, showed: Reticular opacities and honeycomb pattern of subpleural distribution. In addition, the patient presented echocardiographic findings of tricuspid valve flow regurgitation and increased systolic pressure of the right ventricle as indirect measures of PAH. Case number 6 presented three digital ulcers and one on the right foot.
The clinical cases presented came from both rural and urban areas of Guatemala. All were women, who at the time of diagnosis were between the ages of 25 and 71 with a mean of 45, which is consistent with the review of epidemiological studies, as well as the predominance of the female sex [7,29]. The description of MCTD used in clinical cases was based on the criteria of Alarcón-Segovia and Villareal, patients met both clinical and serological criteria, with Anti-RNP values in high titers without the presence of Anti-Sm.
Regarding the initial clinical presentation, we observed heterogeneous signs and symptoms among the patients studied, although the literature generally refers to the most frequent initial symptom: Raynaud's phenomenon, swelling of the hands and arthralgia; In our review we found constitutional symptoms in all cases such as fever and asthenia, followed by: Edema, weight loss, alopecia and sicca symptoms.
All patients had Anti-RNP values greater than 1:1600, without the presence of Anti-Sm. It is important to emphasize that three patients had positive Anti-DNA values, but one of them had borderline values, there was no positivity for Anti-Sm that would be considered more defined as SLE [1,18]. The rest of the studies are more heterogeneous, finding positive RF in all patients in high titres [30], in addition to positivity in two cases for anti SSA/Ro, as well as elevation of total CK in a case that was the only one with myositis. These findings are compatible with the positivity of other antibodies to MCTD, we found no antiphospholipid antibodies or ANCA, as described in the literature in 15-20% [18,31,32].
In Sharp's study, a good response to the use of steroids was observed with a gradual reduction and symptom control scheme, and few cases required the use of more than 10 mg/day of prednisone [2,33]. In our cases at AGAR clinic that required steroids all used less than 10 mg/day of prednisone or equivalent.
In relation to long-term complications, only one patient presented PAH data in addition to ILD, which are classified as the most serious complications of MCTD [34-36]. Although all had Raynaud's phenomenon since the beginning of the disease, only one of them was complicated by digital ulcers in hands and feet that had been described previously [37]. However, each patient should be individualized according to the recommendations of most authors.
MCTD is a well-defined entity, although for many authors it is a preclinical stage for other connective diseases such as SLE, SSc or PM/DM, in our cases the evolution time has even been up to 10 years without modifying the initial diagnosis. The criteria used for the case definition were those of Alarcón-Segovia and Villareal, which are the most simple and specific for diagnosis. The serological profile of the patients is quite heterogeneous, in addition to finding varied constitutional symptoms at the time of diagnosis, as well as hematological alterations. The complications of the disorder have already been described in literature, although only one patient presented complications PAH and ILD, however, it is important to note that both comorbidities worsen patient prognosis. Each case must be individualized, however, because there are no management protocols; management guidelines would be given by the clinical manifestations and the individual profile. Finally it is worth mentioning that in Guatemala there are no previous studies of MCTD.
There were no conflicts of interest from authors.