Cutaneous leishmaniasis (CL) is an infectious disease endemic in tropical and subtropical countries. The current drugs have severe drawbacks that restrict their use and enhance the need for better drugs. Recently, the N-iodomethyl-N,N-dimethyl-N-(6,6-diphenylhex-5-en-1-yl) ammonium iodide (C6I) was identified as a promising compound for the topical treatment of CL. The need for oral drugs with potential use to treat cutaneous, mucosal and visceral leishmaniasis, in the present work were determined the pharmaceutical and some biopharmaceutical properties of C6I as possible oral treatment and based on this results a nanoformulation was elaborated, characterized and tested in in vitro and in vivo model of the antileishmanial activity and toxicological assays. The C6I showed crystalline form and good intestinal permeability, its dissolution profile did not change with pH changes. The C6I was not mutagenic and genotoxic in vitro, it presented some minors acute toxicological effects.
The solid lipid nanoparticles (SLN) used Precirol® as lipid, it had a size in the nano range scale with a low polydispersity index, and encapsulation efficiency > 60%. The nanoparticles of C6I (PC6I) increased the in vitro antileishmanial activity 40-fold than free C6I. In turn, the oral administration of C6I and PC6I (30 mg/kg/d, 28 days) produced complete cure in 42.9% and 71.4%, respectively, with no relapses and no toxicity. The effectiveness of meglumine antimoniate was 100% but the relapse rate was 28.6%. C6I and PC6I are safe compounds, as demonstrated in in vitro and in vivo assays for toxicological profile. In conclusion, a novel oral quaternary ammonium iodide salt-based formulation with antileishmanial properties was developed. The safety and effectiveness information of PC6I formulation showed here supports the further evaluation of efficacy and safety in patients to validate the use of PC6I as an alternative for the oral treatment of CL.
Cutaneous leishmaniasis, Quaternary halomethylated ammonium iodide, Drug development, Solid lipid nanoparticles, Precirol®
Current drug therapies for cutaneous leishmaniasis (CL) are unsatisfactory due to numerous drawbacks including severe side effects, reproductive toxicity, prolonged treatment with high doses and decreased effectiveness. Since CL is prevalent in economically disadvantaged communities, new drugs development lack for economic incentives to large pharmaceutical companies and in consequence, leishmaniasis is designated as neglected tropical disease [1,2].
In a previous work was reported that the quaternary halomethylated ammonium salt N-iodomethyl-N,N-dimethyl-N-(6,6-diphenylhex-5-en-1-yl) ammonium iodide (C6I), a choline analog, could be considered as a lead compound in the development of a new treatment for CL. The C6I has activity against intracellular amastigotes of Leishmania (Viannia) panamensis [3,4]. Moreover, the C6I administered topically at a dose of 2 mg/kg/day during 15 days was able to cure hamsters with experimental CL caused by L. (V) braziliensis [5]. The ability of C6I to inhibit the choline/ethanolamine kinase of L. (Leishmania) infantum and the production of phosphatidyl choline and to interfere with the in vitro uptake of choline by promastigotes of L. tarentolae was also demonstrated [6]. The organoleptic analyses showed that C6I is an odorless, white-colored powder with fine appearance, soft to the touch, crystalline habits with regular plaque shapes and smooth texture, grouped in clusters and particle size varying between 20 and 100 microns. Other physiochemical studies showed that the solubility of C6I is high in acetonitrile (1.5 mg/mL), low in methanol, isopropanol and chloroform (< 1.0 mg/mL) and water (0.61 mg/mL). This solubility is not affected by pH changes [5].
The importance of C6I is higher when considering the urgency expressed by the World Health Organization of having drugs for oral administration since miltefosine, the only medicine available for oral treatment of leishmaniasis, is not widely available in the market.
In the present work were determined the pharmaceutical properties of C6I as well as the usefulness of oral C6I to cure experimental CL in hamsters. The in vitro cytotoxicity, mutagenicity and genotoxicity of C6I were also determined. Because the low solubility observed previously for C6I could difficult the dissolution process in the GI tract and, affect the bioavailability [7], the oral delivery can be improved using nanocarrier systems such as solid lipid nanoparticles (SLNs) that are at the forefront of the potential application mainly due to the low cost and easy industrial production [8]. Thus, in the present work were prepared and characterized SLNs of C6I using Precirol® (PC6I). The in vitro and in vivo antileishmanial toxicity and activity of PC6I were assessed against L. (V) braziliensis, compared to the antileishmanial activity of free C6I and with the standard treatment to this disease, meglumine antimoniate (MA). Finally, the in vivo oral toxicity profile was also determined.
The C6I was supplied by L.A Rios and R. Ocampo. Its synthesis was previously described by Duque, et al. 2016 [3].
The study for intestinal absorption was approved by the Ethical Committee for Animal Welfare of the University of Valencia (Act No. CE 86/609) while the in vivo assays for, in vivo effectiveness and toxicity, maximum tolerable dose and oral toxicity at repeated dose were approved by the Ethical Committee for Animal Experimentation of the Universidad de Antioquia (Act No. 91-2014).
One hundred mg of C6I were compressed at 700 Pa and a 6.33 mm punch during 1 min. The surface area of the compacts was 3.14 cm2 [9]. Compacts were placed in a molten beeswax-mold with one face in contact with dissolution medium. Dissolution was conducted in an USP type 2 dissolution apparatus using 900 mL of either 0.1 N HCl, 0.2 M acetate buffer, pH 1.5, purified water pH 7.0 and 0.2 M phosphate buffer, pH 8.0 at 37 ± 1 ℃, as dissolution media, respectively, with a paddle rotating at 100 rpm. One mL samples were withdrawn at regular intervals and a plot of absorbance vs. time was constructed.
Experiments were done in triplicate. Absorbance was determined using the UV-Vis spectrophotometer (VarioScan Thermo) at 233. The dissolved cumulative amount per surface unit of the compact was plotted against time for each vessel. A standard curve was built using standard solutions used in dissolution media. Intrinsic dissolution rate (IDR) was calculated using Eq. 1:
Where j is the IDR, V is the volume of the dissolution medium, c is the concentration, A is the area of the C6I disk and t is the time
The melting points, purity and crystalline appearance of C6I were determined by differential scanning calorimetry (DSC) profile using a DSC 204 F1 (Phoenix®-NETZSCH-Gerätebau GmbH). Measurements were in a range of 25 ℃ to 300 ℃ with increases in 10 ℃ per min. The temperature scale was calibrated using an internal reference within pots of aluminum. Tests were performed in a nitrogen atmosphere.
The crystallinity properties of C6I were determined by Powder x-ray diffraction (PXRD) using an X-ray diffractometer Rigaku Miniflex® (Rigaku, Tokyo, Japan) with a source of Cu, Ka1 radiation (1.542 A°) and angles ranging between 3° and 50°.
Thirteen adult Wistar male rats were fasted for 18 h (except for drinking) and then anesthetized and immobilized. Afterwards, the abdominal cavity was cut along the medioventral line, and the small intestine was separated. Then, the small intestine was washed with 0.9% saline solution and two different concentration of compound was used, 10 mL of supersaturated or 1/10 diluted C6I solution were perfused. The concentration of C6I in the intestinal lumen was measured every 5 min for 30 min by sampling 200 µL of the remaining liquid. Samples were immediately centrifuged (1,500 g, 10 min) and the supernatant frozen (-80 ℃) until quantification by HPLC using the method described [5]. Samples of 1.0 mg/mL of C6I were prepared and run in an Agilent Technologies 1200 chromatograph equipped with a C18 column (Restek C18, 5 µm, 250 × 4.6 mm), with UV-detector. The mobile phase consisted of acetonitrile and C2H3NaO2 0.1 M (60:40, v/v), adjusted to pH 5.0 with acetic acid. Elution was achieved in 12 min at a flow rate of 0.8 mL/min. The injection volume was 10 µL. Compound was monitored at 233 nm. At the end of the sampling, the remaining volume was recovered and the extent of water reabsorption was determined at every sampling time, considering that this process follows zero-order kinetics [10]. The apparent constant absorption rate for C6I was obtained from the first-order equation Eq. 2:
Where C is the concentration of C6I remaining in the intestinal lumen, ka is the apparent rate constant for absorption, and C0 is the initial concentration of C6I.
The ka was transformed in to permeability values by means of Eq. 3:
Where R is the effective radius of the intestinal segment corresponding to 0.1784 cm, considering that intestinal segment as a cylinder of 10 mL volume and 100 cm length.
Salmonella typhimurium TA98 and TA100 adjusted at 2 × 109 CFU/mL and 100-μL were added to assay tubes containing C6I (46, 23 and 12 µg/mL) and 500 μL of phosphate buffer (Na2HPO4 and NaH2PO4·H2O) - system without metabolic activation -, and pre-incubated at 37 ℃ for 30 min. Sodium azide (AzNa) or 4-Nitroquinoline (4NQO) (5 µg/mL) were used as positive control for S. typhimurium TA100 and TA98 strain, respectively. Phosphate buffer was used as negative control. Revertants (his+) were counted after incubation at 37 ℃ for 66 h [11].
Chromosome alterations induced by C6I were evaluated in lymphocytes from three healthy donors [12]. Cells cultured in RPMI 1640 medium supplemented with 2 mM L-glutamine, 10% fetal bovine serum and 1% penicillin-streptomycin (10,000 U-10 mg/mL) (complete medium) were stimulated with 200 µL phytohemagglutinin and incubated at 37 ℃, 5% CO2. After 36 hours, the cells were treated with each of sublethal C6I concentrations (46 - 23 and 12 µg/mL) and incubated during 24 h. Colcemid (0.1 pg/ml) was added and cells were incubated again during 1 h. The cells were centrifuged, resuspended in 0.075 M KCl and incubated at 37 ℃ for 7 min, fixed 10 min with Carnoy solution, washed and dripping. Slides were dried and stained with 5% Giemsa, pH 6.8. One hundred metaphase cells (46 chromosomes) were analyzed. Structural chromosome aberrations were classified as chromatid breaks (B), chromosome breaks (BB), dicentric chromosomes (DC), ring chromosomes (R) and multiradial chromosomes (MR). Each treatment was performed in two separate experiments using RPMI as negative control and mitomycin-C (10 M) as positive control.
Twenty-one female, nulliparous and non-pregnant Wistar rats, 188 ± 12 g weight, were divided into four groups. Three groups (n = 6 each) were treated with a single dose of 5, 50 or 300 mg/kg (1 mL) of C6I by the oral route. The forth group (n = 3) received distilled water (control group). Rats were observed during the first 6 h for any potential signs of toxicity and then daily during 14 days. Signs were graded according their nature and severity. Rats were weighed before and every week after doses. At day 14 rats were sacrificed and necropsied. The heart, brain, liver, spleen, kidneys, testicles and epididymis (males) and ovaries (females) were weighed and biopsies were stored in 10% formaldehyde for histopathology analysis [13]. The severity of lesions was graded from 0 to 3, where 0 for no lesion, mild, moderate or severe, respectively. The surviving rats were monitored for mortality, behavior, fur, pain and any sign of illness during the study.
The Precirol® lipid was selected because it is frequently used in oral formulation, the production method of SLN was selected, first to determine of solubility of C6I in Precirol®, one milligram of compound was added to 25 mg of lipid that was heated above 10 °C its melting points. Solubility was verified after that the excess of C6I persisted for more than 8 hours [14].
The Precirol® NPs loaded with C6I (PC6I) were elaborated by evaporation of the emulsion-solvent using 0.6% sodium deoxycholate, 0.5% Tween 20 and 1% soy lecithin as co-surfactants [15]. Fifty mg of Precirol® was dissolved with lecithin and C6I (3 mg) in 1 ml of dichloromethane. This organic phase was then added to 5 mL of aqueous phase containing Tween 20 and sodium deoxycholate. Dispersion was performed by sonication for 3 min (Branson Sonifier 250, USA) followed by 5 min of stirring at 12,500 rpm for 4 h at room temperature using a High Speed Mixer (Model L4RT, Silverson Machines, Chesham, UK) until the dichloromethane had completely evaporated. Free C6I was separated from the dispersed Precirol® NPs by size exclusion chromatography in a PD-10 column (Bio-Rad Laboratories) using as eluent phosphate buffer saline (PBS), pH 7.4.
The NPs were characterized based on particle size and surface charge (PZs) determined by photon correlation spectroscopy (PCS) using a Zetasizer Nano S (Malvern Instruments, UK). Measurements of particle size were performed at 25.0 ± 0.1 ℃ after dilution of the NPs in purified water (1:100). The results were expressed as the average of particle size and the polydispersion index (PI). In turn, the surface PZ value of the NPs was determined based on the mobility of the particles in an electric field using a Zetasizer Nano Z. For this, the diluted samples in purified water were exposed to 150 mV and the appearance.
The incorporation of C6I into the NPs was quantified after dissolving the NPs loaded with C6I in acetonitrile, which promoted precipitation of the lipid phase. After centrifuging, free C6I was measured at 233 nm in a microplate spectrophotometer (FLUOstar Omega). The supernatant from unloaded NPs was used as a blank. The quantification method was validated according to the international guide for validation of analytical procedures [16]. The efficiencies of encapsulation (EE) of C6I and drug loading (DL) in the NPs were calculated according to Eq. 4 and Eq. 5:
The morphology of the NPs were determined by scanning electron microscopy (SEM, XL-30 Royal Philips Electronics, Amsterdam). All measurements were performed in triplicate.
stored at 2-8 ℃, and the mean particle diameter, PI, zeta potential and DL were determined after 30 and 60 days of storage. The average of particle size was analyzed as previously described. The stability evaluation in terms of EE and DL was determined after separation of unbound C6I by size exclusion chromatography, as described above. The effect of autoclaving was also evaluated, for this assessment, the PC6I formulation was divided into two aliquots of equal volume. One aliquot was autoclaved at 121 ℃ for 20 min, while the other aliquot was maintained at 2-8 ℃ as a reference sample and then, the physicochemical properties (particle diameter, PI, surface charge, DL and EE) were compared. Additionally, the influence of temperature on the physical stability of PC6I was evaluated using DLS (Zetasizer Nano S). Samples were diluted with purified water (1:100) in a quartz cell, and particle size analysis was performed while heating the sample from 25 ℃ to 90 ℃ at a rate of 0.5 ℃/min and then cooling it from 90 ℃ to 25 ℃ at the same rate. Particle size was measured every 0.5 ℃. This assay was done in triplicated.
The NPs were incubated in 10 mM PBS release medium, pH 7.4, with horizontal shaking at 37 ℃. At 0.5 - 1 - 2 - 4 - 8 and 24 h intervals samples were centrifuged at 30,000 × g for 30 min at 4 ℃. The released C6I was quantified in the supernatants by spectrophotometry in a UV-1700 microplate reader at 233 nm. Quantifications were done in triplicate [15].
The DSC measurements were performed on a DSC calorimeter Q200 (TA Instruments, DE, USA). Dispersions of C6I, Precirol®, sodium deoxycholate, Tween 20, soy lecithin, empty NPs and PC6I nanoparticles were weighed and measured against an empty reference vessel. The samples were heated, and the respective thermograms were recorded in the temperature range of -20 ℃ to 240 ℃ at a heating rate of 10 ℃/ min.
The cytotoxicity of free C6I, empty NPs and PC6I was evaluated in vitro on primary cultures of human monocyte-derived macrophages (MDMhu) and golden hamster peritoneal macrophages (MPha) and human cell lines U937 (ATCC® CRL-1593.2™), Caco-2 (ATCC® HTB-37™) and Detroit 551 (ATCC® CCL-110™). Cytotoxicity was determined by cell viability using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) enzymatic micromethod following a methodology described by others [17]. Untreated cells were used as a viability control and doxorubicin was used as a positive control for cytotoxicity. At least two independent experiments were performed, each in triplicate. The % of mortality was used to calculate the half maximal lethal concentration (LC50) using the Probit analysis [18].
Intracellular amastigotes of L. (V) braziliensis (MHOM/CO/88/UA301-EGFP) were obtained after infection of U937 cells as described elsewhere [19]. After 24 h of infection the medium was replaced by fresh medium containing each C6I, empty NPs or PC6I, at concentrations of 100 - 12.5 - 3.125 - 0.78 µg/μL. After 72 h, cells infected and treated were analyzed by flow cytometry (FC 500MPL, Cytomics, Brea, CA, USA). The antileishmanial activity was determined according to the reduction in the number of viable parasites inside the infected cells obtained with each treatment and each concentration, according to the mean fluorescence intensity (MFI). Data obtained in untreated infected cells corresponds to 100% of parasites. All measurements were performed in triplicate, in two independent experiments. The half maximal effective concentration (EC50) was calculated according to the Probit analysis [18].
Male and female hamsters, 6-weeks old, were inoculated in the dorsum with 5 × 108 promastigotes of L. (V) braziliensis (MHOM/CO/88/UA301-EGFP)/100 mL. When the ulcer was developed in the skin, hamsters were distributed in five experimental groups. Two group of hamsters (n = 7 each) received by oral route 1 mL of C6I or PC6I at a dose of 30 mg/kg/day for 28 days. A third group (n = 7) was treated with MA at a dose of 120 mg/kg/day for 10 days, by intramuscular route, that corresponds to the curative dose identified in previous work (cure control) [20]. A fourth group (n = 3) was treated with the empty NPs (vehicle) and the fifth group (n = 3) remained untreated after infected (non healing control).
The clinical effectiveness of each treatment was determined by comparing the lesion sizes before treatment (TD0) with those observed the last day of treatment (TD28 for C6I, PC6I and NPs), or TD10 for MA) and days 30, 60 and 90 of follow-up after treatment (PTD30, PTD60 and PTD90, respectively). At the end of the study that is PDT90, the outcome was recorded as "cure" (complete disappearance of the lesion), "improvement" (more than 20% of decrease of the area), "failure" (increasing of the area), or "relapse" (reactivation of lesion after an initial cure). The parasitological effectiveness was also determined by quantification of parasite load in the skin samples from the ulcer at the end of the study (PTD90) by RT-PCR, following the protocol described by others [21].
The toxicity of C6I, PC6I, NPs and MA was studied based on changes in the weight of hamsters measured every two weeks previous sedation. Additionally, at TD0 and day 8 after the end of treatment (PTD8) hamsters were bled by cardiac punction and serum was separated by centrifuging (5 min at 5,000 × g). Levels of alanine amino transferase (ALT), blood urea nitrogen (BUN) and creatinine (CRT) metabolites were quantified using commercial kits (Biosystems S.A, Barcelona, Spain). Hamsters were monitored daily for food and water consumption, activity and behavior. The appearance of fur, eyes and mucous membranes was also supervised.
Fifty healthy white Swiss Webster mice (CFW) of 18-22 g weight were divided into five groups (five males and five females, each). Two groups received 0.2 mL of C6I or PC6I at 30 mg/kg/day, respectively by oral route during 28 days. A third group remained untreated (negative control). Mice were monitored daily and weighted before treatment and weekly during treatment. Blood samples were obtained prior to the first dose (TD0) and at the end of the study (TD28) [22]. At the end of the study animals were sacrificed and necropsy and biopsies were performed. Serum level of aspartic transaminase (AST), ALT, BUN, CRT, albumin (ALB) and alkaline phosphatase (AP) were quantified. The blood cell parameters were also evaluated.
Data are expressed as mean ± SD, except for effectiveness of treatments that are expressed as percentage. The Kolmogorov-Smirnov test was used for normality distribution of data and differences between variables were analyzed by Student’s t-test using GraphPad Prism version 6.0 (GraphPad Software, San Diego, CA, USA). Differences were considered significant when p < 0.05.
Intrinsic dissolution of pure compounds did not change with different pHs, the value of intrinsic rates (IDR) were similar in tree conditions, 0.24 ± 0.03 at pH 1.5; 0.26 ± 0.04 at pH 7.0 and 0.20 ± 0.05 at pH 8.0 (mg/min/cm2).
The DSC and powder X-ray diffractometric data are summarized in Figure 1. The C6I had a melting point of 160.1 ℃, an exothermic peak close to 150 ℃, possibly due to the sublimation of iodine (Figure 1A) and an internal structure predominantly was crystalline confirmed by the XRPD analysis (Figure 1B).
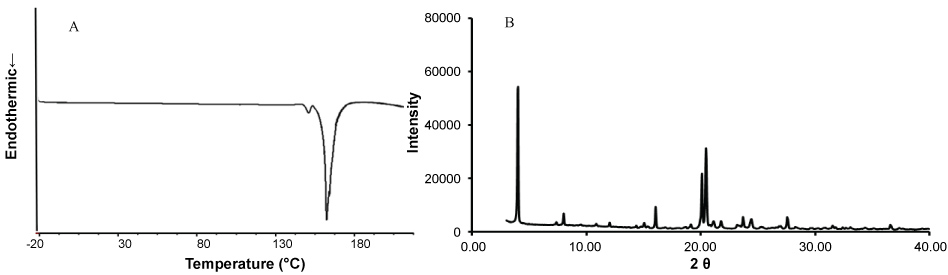 Figure 1: Thermogram and X-ray diffraction of quaternary halomethylated ammonium iodide (C6I). (A) DCS profile, exothermic peak to 160.1 ℃ corresponds to the melting point of C6I; (B) XRPD profiles of C6I shows an internal structure predominantly crystalline.
View Figure 1
Figure 1: Thermogram and X-ray diffraction of quaternary halomethylated ammonium iodide (C6I). (A) DCS profile, exothermic peak to 160.1 ℃ corresponds to the melting point of C6I; (B) XRPD profiles of C6I shows an internal structure predominantly crystalline.
View Figure 1
The individual absorption rate constant (ka) and Papp permeability values of C6I obtained using a saturated solution and a 1/10 dilution solution were similar value without statistically significant differences, the permeability process in intestinal wall was independent of concentration (p > 0.05) (Table 1).
Table 1: Intestinal permeability of quaternary halomethylated ammonium iodide (C6I). View Table 1
There was no significant increase in the number of revertant colonies of the TA98 and TA100 S. typhimirium strains in the three concentrations of C6I compared to the negative controls. The test was done without exogenous metabolic activation system (p > 0.05). The positive controls AzNa and 4-NQO increased two times in the mutagenicity index compared to C6I and negative control.
None structural chromosome aberrations were identified in the different concentrations of C6I evaluated. No statistically significant differences were found between the subtoxic concentrations of C6I and the negative control.
During the 14 days of observation after oral administration of three different concentrations of C6I (5, 50 or 300 mg/kg), there were no deaths or evidence of clinical signs associated with severe toxicity or changes in the normal behavior of rats. A soft but not diarrheic stool was observed a day after the compound C6I was administered but it was normalized during the same day. A weight gain was observed in rats of all groups. There was no evidence of pathological signs during necropsy and no histological changes were confirmed. Therefore, it is possible to administer at one time a maximum concentration of pure compound of 300 mg/kg without significant effects.
The C6I was not soluble in Precirol® after fusion. Therefore, the formulation of PC6I was prepared by emulsion and solvent evaporation processes. The particle size was homogeneous and this was range of nanometer, the encapsulation performance was 68% (2.04 mg of C6I ), and the PI values was lower than 0.3 (Table 2).
Table 2: Physicochemical properties and stability of quaternary halomethylated ammonium iodide incorporated in Precirol® nanoparticles. View Table 2
The SEM analyses showed that the NPs had spherical and homogeneous shape and some aggregates were present (Figure 2).
 Figure 2: SEM images of quaternary halomethylated ammonium iodide incorporated in Precirol® nanoparticles (PC6I). A) 2,350X. B) 7,000X. Notice of Spherical shape of nanoparticles.
View Figure 2
Figure 2: SEM images of quaternary halomethylated ammonium iodide incorporated in Precirol® nanoparticles (PC6I). A) 2,350X. B) 7,000X. Notice of Spherical shape of nanoparticles.
View Figure 2
Storage of the NPs under refrigeration condition generated a slight decrease of the particle size of the NPs during the first month. On the other hand, the zeta potential increased in the first month but decreased in the second month. Unloaded NPs were not affected at the same extent by extensive refrigeration.
The heating processes by autoclaving did affect the particle sizes and the zeta potential decreased (Table 2). Additionally, DLS analysis showed that by increasing the temperature at 90 ℃ there was a gradual reduction in the particle size. Although the original size was not recovered after the cooling phase (Figure 3A), the heating and cooling process did not affect significantly of size of particles the values were always within the nano range.
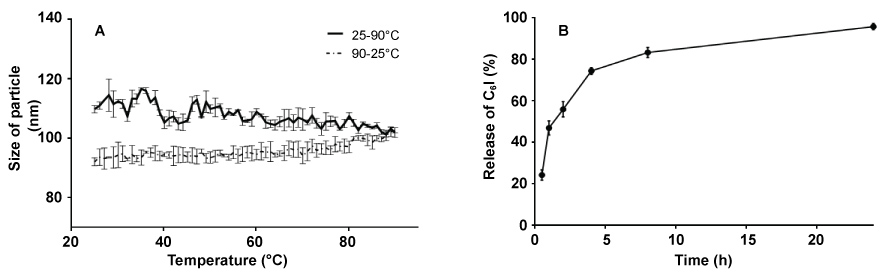 Figure 3: Release of quaternary halomethylated ammonium iodide from Precirol® nanoparticles in vitro and thermal analysis. (A) DLS thermogram of quaternary halomethylated ammonium iodide incorporated in precirol nanoparticles (PC6I); (B) Release profile in 10 mM PBS pH 7.4 at 37 ℃. data represent the mean value ± SD, n = 3. View Figure 3
Figure 3: Release of quaternary halomethylated ammonium iodide from Precirol® nanoparticles in vitro and thermal analysis. (A) DLS thermogram of quaternary halomethylated ammonium iodide incorporated in precirol nanoparticles (PC6I); (B) Release profile in 10 mM PBS pH 7.4 at 37 ℃. data represent the mean value ± SD, n = 3. View Figure 3
There was a rapid release phase in the first 30 min followed by an exponential phase of release in the following 22 hours (Figure 3B). The efficiency of release of C6I was ≥ 94% after 20 hours of observation. The quantification method for C6I showed a sensitivity and accuracy according to the concentration range. The linearity varied from 0.03875 to 0.6 mg/mL, with a SD of 0.08 and a coefficient of variation of 3.1%. The limit of quantification (LOQ) and the limit of detection (LOD) were 0.03 and 0.009 mg/mL, respectively, with a confidence interval of 99.25%.
The melting point (mp) of Precirol® and C6I were 70-80 ℃ and 163 ℃, respectively. However, in the PC6I, the melting point peak corresponding to C6I was absent, possibly due to a structural change of the compound within the NPs from its crystalline form to an amorphous or molecularly dispersed state or the concentration of the compound is lower than of the lipid, which did not allow looking its peak. The Figure 4 shows the thermograms of each of the components of formulation, unloaded NPs and PC6I.
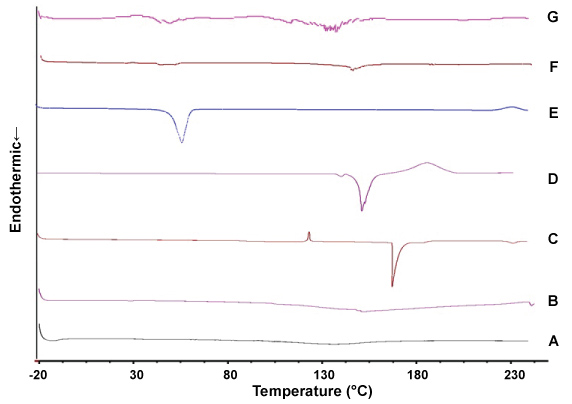 Figure 4: Thermograms of the components of Precirol® nanoparticles. (A) Tween 20, (B) lecithin, (C) sodium deoxycholate, (D) free C6I, (E) Precirol®, (F) empty Precirol® nanoparticles, (G) Precirol® NP with C6I. View Figure 4
Figure 4: Thermograms of the components of Precirol® nanoparticles. (A) Tween 20, (B) lecithin, (C) sodium deoxycholate, (D) free C6I, (E) Precirol®, (F) empty Precirol® nanoparticles, (G) Precirol® NP with C6I. View Figure 4
To compare the results of free C6I and encapsulated, first, the cytotoxic effects on fibroblast (Detroit 551) and MDMhu the doses was LC50 < 100 μg/mL for C6I while PC6I was not cytotoxic for all the cell types evaluated, with a LC50 > 200 μg/mL (Table 3). In relation with the effectivity, C6I had an EC50 17.6 ± 1.0 μg/mL while PC6I showed an EC50 of 0.4 ± 0.2. The ratio of cytotoxicity and activity (LC50/EC50) showed an IS of 0.53 and higher than 500 μg/mL for C6I and PC6I, respectively. These results suggest that the selectivity of C6I for the parasite increased significantly when was incorporated into the Precirol® NPs (Table 3).
Table 3: in vitro cytotoxicity and antileishmanial activity of free quaternary halomethylated ammonium iodide and incorporated in nanoparticles. View Table 3
After experimental infection, hamsters developed a single lesion ranged from 113.5 to 199.3 mm2. The treatment of hamsters with free C6I, administered orally at a dose of 30 mg/kg/day for 28 days, resulted in complete healing of ulcers in 3 animals (42.9%) at the end of the study (Figure 5). The remained hamsters that did not cure showed improvement with a reduction in the size of their lesions in 90.4, 38.4, 37.05 and 8.6%. On the other hand, the treatment of hamsters with PC6I, administered orally at the same dose during 28 days increased the cure rate to 85.7% corresponding to 6 of 7 hamsters with complete healing of ulcers. The remained hamster of this group showed a decrease in the lesion of 95.9% (hamster PC6I-5, Figure 5). One of cured hamster had relapse at the end of the study (hamster PC6I-7, Figure 5) for a definitive cure rate of 71.4% for PC6I treatment. Although all hamsters treated with intramuscular injections of MA showed cure during the study, only 5/7 (71.4%) hamsters remained cured at the end of the study while the last two hamsters had relapses of their ulcers. As expected, any hamster from the vehicle treated or untreated groups resolved their lesions during the study (Figure 5A). The appearance of lesion in hamsters prior and after treatment with C6I and PC6I and at the end of the study are shown in Figure 5B, Figure 6.
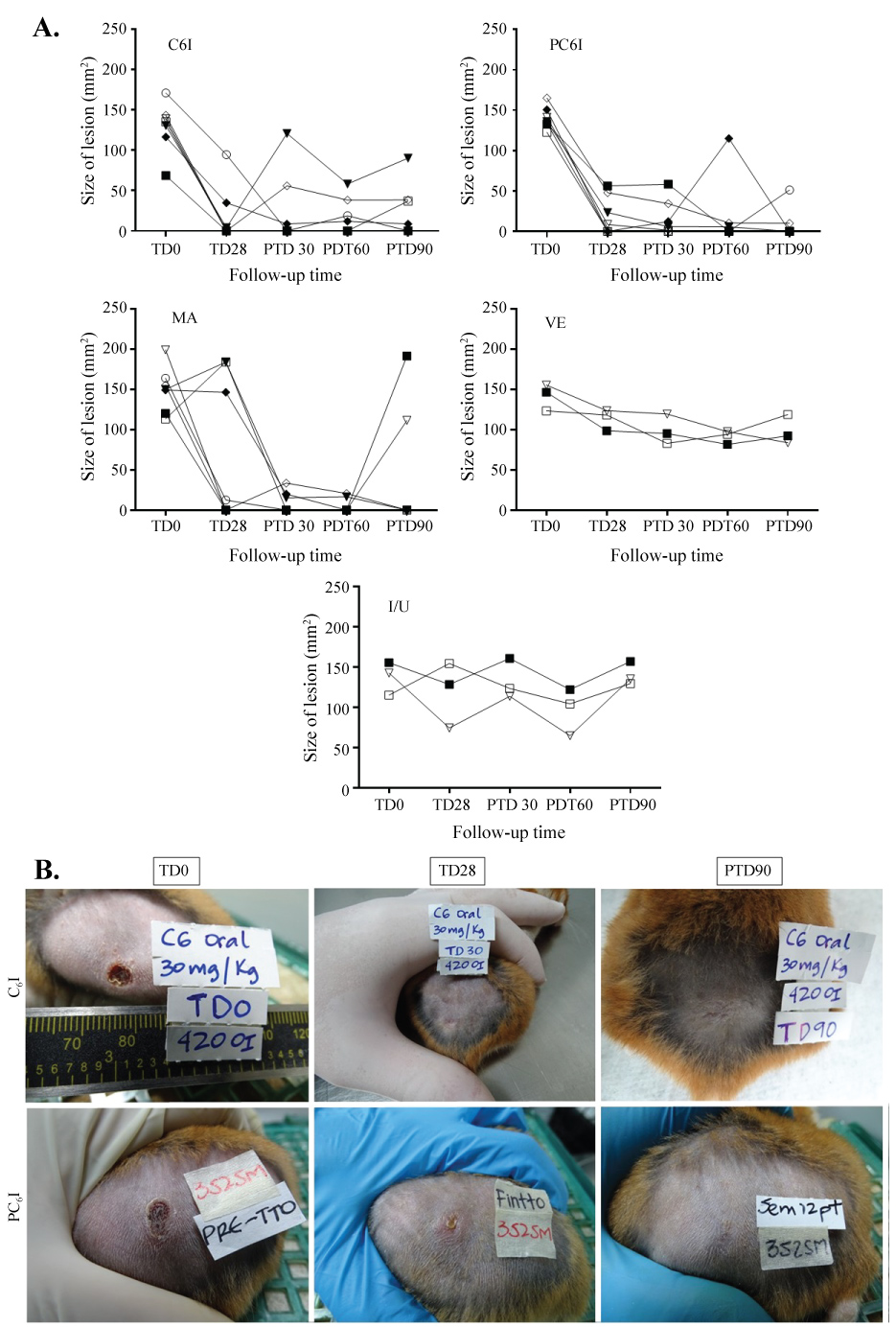 Figure 5: Response of hamsters after treatment with free quaternary halomethylated ammonium iodide and incorporated in Precirol® nanoparticules vs. meglumine antimoniate. (A) The graphs show the outcomes of hamsters after treatment with oral free C6I, Precirol® NP with C6I (PC6I), intramuscular meglumine antimoniate (MA) or vehicle (VE) in comparison with infected and untreated hamsters. Evaluations were done at the end of treatment and post-treatment days 30 (PTD30), 60 (PTD60) and 90 (PTD90). Data represent the size of lesions at each time of follow-up. (B) Treatment progress of cutaneous leishmaniasis caused by L. (V) braziliensis in one hamster treated with C6I and one treated with PC6I (a) Before treatment (TD0); (b) On day 28 at the end of treatment (TD28) and (c) on day 90 post treatment (PTD90). View Figure 5
Figure 5: Response of hamsters after treatment with free quaternary halomethylated ammonium iodide and incorporated in Precirol® nanoparticules vs. meglumine antimoniate. (A) The graphs show the outcomes of hamsters after treatment with oral free C6I, Precirol® NP with C6I (PC6I), intramuscular meglumine antimoniate (MA) or vehicle (VE) in comparison with infected and untreated hamsters. Evaluations were done at the end of treatment and post-treatment days 30 (PTD30), 60 (PTD60) and 90 (PTD90). Data represent the size of lesions at each time of follow-up. (B) Treatment progress of cutaneous leishmaniasis caused by L. (V) braziliensis in one hamster treated with C6I and one treated with PC6I (a) Before treatment (TD0); (b) On day 28 at the end of treatment (TD28) and (c) on day 90 post treatment (PTD90). View Figure 5
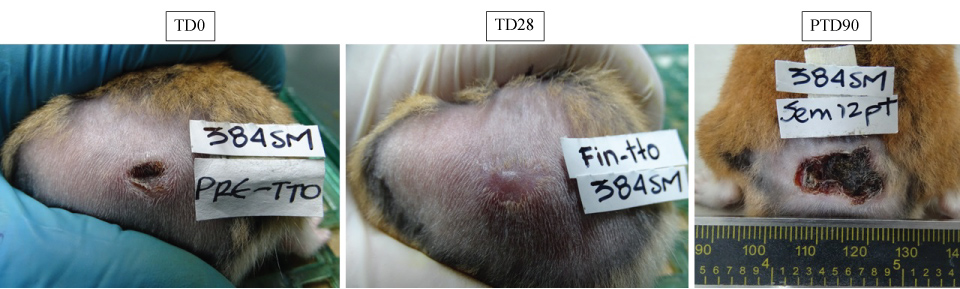 Figure 6: in vivo effect of free quaternary halomethylated ammonium iodide and incorporated in Precirol® nanoparticules. The figure shows representative images of a skin injury caused after injection of 5 × 107 by Leishmania (V) braziliensis in one hamsters and their evolution after treatment with PC6I at 30 mg/kg/day during 28 days. (a) appearance of lesion before treatment (TD0); (b) appearance of lesion at the end of treatment (TD28) and (c) appearance of skin 90 days after treatment ended (PTD90). Notice the decrease in size after treatment and the development of scar tissue after cure. View Figure 6
Figure 6: in vivo effect of free quaternary halomethylated ammonium iodide and incorporated in Precirol® nanoparticules. The figure shows representative images of a skin injury caused after injection of 5 × 107 by Leishmania (V) braziliensis in one hamsters and their evolution after treatment with PC6I at 30 mg/kg/day during 28 days. (a) appearance of lesion before treatment (TD0); (b) appearance of lesion at the end of treatment (TD28) and (c) appearance of skin 90 days after treatment ended (PTD90). Notice the decrease in size after treatment and the development of scar tissue after cure. View Figure 6
The parasite load expressed as at the end of the study in the ulcers by group of treatment was 644.4 ± 953.8 parasites/mg of tissue (mean ± SD, n = 7) and 303.2 ± 288.6 parasites/mg of tissue in hamsters treated with C6I and PC6I, respectively. These difference was statistically significant (p < 0.01).
The hamsters gained weight during the study and no statistically significant differences were observed among groups (Figure 7). Serum levels for all animals of ALT, BUN, and CRT were within the range of normal values for all animals both before (TD0) and after (PTD8) the all treatments (Figure 8).
 Figure 7: Effect of treatment in the weight of hamsters. The figure shows the evolution of the weight of hamsters in each group of treatment. Data represents the mean value ± SD of the weight in grams of hamsters in each experimental group: oral free C6I; Oral Precirol® NP with C6I (PC6I); Intramuscular meglumine antimoniate (MA), Vehicle (VE) or Untreated (I/U). View Figure 7
Figure 7: Effect of treatment in the weight of hamsters. The figure shows the evolution of the weight of hamsters in each group of treatment. Data represents the mean value ± SD of the weight in grams of hamsters in each experimental group: oral free C6I; Oral Precirol® NP with C6I (PC6I); Intramuscular meglumine antimoniate (MA), Vehicle (VE) or Untreated (I/U). View Figure 7
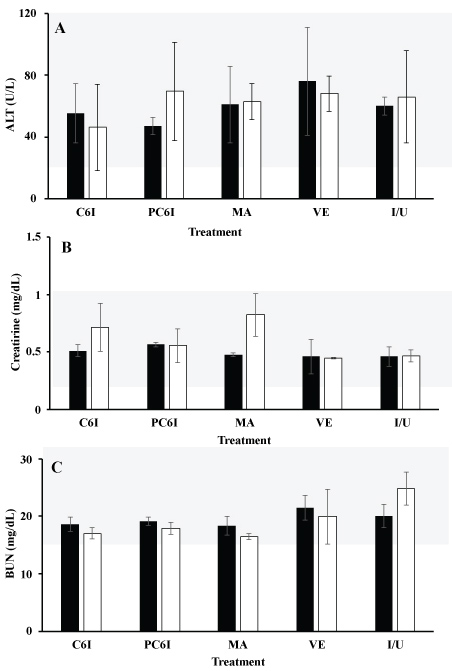 Figure 8: Comparison of BUN, creatinine and ALT Level in serum of hamsters treated with free quaternary halomethylated ammonium iodide and incorporated in Precirol® nanoparticules vs. meglumine antimoniate. (a) Comparison of ALT BUN (b) and creatinine (c) in serum of treatment groups before (TD0) and 8 days after treatment ended (PTD8) in hamsters with cutaneous leishmaniasis. Data are shown as median ± SD. No significant differences were seen between groups (p > 0.05). Precirol® nanoparticles incorporating C6I (PC6I); meglumine antimoniate (MA); Empty Precirol® nanoparticles (VE); Infected and untreated hamsters (I/U). No differences were observed neither among individuals in each group nor between groups. Dotted lines in red delimit the reference values. View Figure 8
Figure 8: Comparison of BUN, creatinine and ALT Level in serum of hamsters treated with free quaternary halomethylated ammonium iodide and incorporated in Precirol® nanoparticules vs. meglumine antimoniate. (a) Comparison of ALT BUN (b) and creatinine (c) in serum of treatment groups before (TD0) and 8 days after treatment ended (PTD8) in hamsters with cutaneous leishmaniasis. Data are shown as median ± SD. No significant differences were seen between groups (p > 0.05). Precirol® nanoparticles incorporating C6I (PC6I); meglumine antimoniate (MA); Empty Precirol® nanoparticles (VE); Infected and untreated hamsters (I/U). No differences were observed neither among individuals in each group nor between groups. Dotted lines in red delimit the reference values. View Figure 8
No deaths or disabling events were neither registered nor clinical signs associated with toxicity of the compounds. The behavior of the mice and their physical appearance were normal even after the administration of the substances. Mice were attentive to the medium, exploring in the cages, grooming each other or adopting typical positions to rest or sleep. Mice treated with free C6I at 30 mg/kg/day showed alterations at the gastrointestinal level with soft stools from the middle of the study to the end without affect the body weight. At contrary, at the end of the study all treated with free C6I or PC6I presented weight gain. At necropsy, only hepatic liver-type changes were reported in two animals of the PC6I group. At the histological level, slight vacuolar degeneration was observed in the kidney, liver and small intestine while slight hyperplasia was observed in liver and spleen of some hamsters treated with both C6I and PC6I. The hematological parameters and serum levels of ALT, AST, CRT and BUN were within the reference values indicating no alterations in the kidney and liver function for both treatments.
One of the objectives pursued for the treatment of leishmaniasis is the development of more effective and safer active pharmaceutical ingredients that can be administered topical or orally to ensure greater acceptance by patients. Previously studies of the quaternary halomethylated ammonium iodide C6I meets the requirements mentioned previously. Our study continued with the pharmacological characterization; the C6I exhibited low dissolution profile with a IDR value similar in the three different pH, additionally the DSC and DRX analysis confirmed that C6I presented a crystalline structure, as described by others authors [7,8]. This crystalline lattice is mainly due to the presence of aromatic rings, which tend to form strong and organize crystalline structure, compounds with this type of structure as usual they present low solubility and dissolution [8,9].
The Doluisio method showed good intestinal permeability of C6I, with Papp one order of magnitude greater than that showed by metoprolol (5.6 × 10-6 cm/s) and in the same range of ibuprofen, which are drugs with good intestinal permeability [10]. On the other hand, rate constants (ka) were the first order, which showed the intestinal absorption of C6I was a passive diffusion process, since there were no statistically significant differences between the constants of the two concentrations tested [23]. The passive diffusion of C6I is favored by its small molecular weight, positive charge and moderate polarity. Some authors report that compounds with positive charge and molecular weight below 400 Da could access at intestinal epithelium through paracellular pathway [24,25]. Together these results and those reported previously [5] allowed to classify the C6I as type 2 compound according to the biopharmaceutical classification system (BCS), with low solubility and good intestinal bioavailability. In turn, the bioavailability of type 2 compounds can affect the amount of drug in solution as well as the amount of C6I that can be absorbed at gastrointestinal level; therefore, type 2 compounds should adapt new strategies to improve their gastrointestinal biopharmaceutical profile, with the purpose to increase the amount of compound bioavailable at systemic level [26].
In acute and repeated doses were evidenced slight edema, vascular congestion and vacuolar degeneration. Despite these signs, organ size and serum metabolites related to hepatic function were normal. On the other hand, C6I did not show mutagenic activity in the S. typhimurium model (TA98 and TA100 strains) and nor chromosomal alterations in human lymphocytes. These results are similar to those showed by other quaternary ammonium compounds [27].
In this work, we use Precirol® not only because it allows the manufacture of NPs easily and economically and by storage conditions, but mainly because it allows the controlled release of drugs [8,28]. The Precirol® NPs improved the pharmaceutical profile (higher effectiveness and lower toxicity) and the biopharmaceutical profile (bioavailability) for C6I as demonstrated by others [29,30].
The C6I was solubilized in a small amount of dichloromethane before the incorporation in the lipid phase. Using 3 mg of C6I, the percentage of encapsulation was higher than 60%. Although the zeta potential was outside the desired range (+30 to -30) [31], the obtained values still indicate good colloidal stability, considering the steric stabilization given by the Tween 20 used as surfactant [32].
The stability tested showed, the storage condition, autoclave conditions and DLS test did not change significantly the mainly characteristics of formulation as size and encapsulation capacity. Authors reported the SLNs as stable systems in different conditions [33]. In contrast, the zeta potential was significantly lower which may be due to a reorganization of the polymorphic transitions of the lipid matrices with subsequent rearrangement of the compound within the formulation [8,28,29].
The DSC analyses showed a rearrangement of the particles compared with the bulk product. The change in the DSC profile could be due to possible defects in the lipid network, leading to a decrease in their crystallinity compared with their bulk counterparts [34]. Thus, for less ordered or amorphous crystals, the fusion of the substance requires much less energy than that for crystalline substances. Additionally, the crystallization processes during the production are complex and strongly dependent on the lipid composition and production conditions as they directly affect the location of the active compound within the particle and its release profile [35]. After 24 hours the 96% of C6I was released, being exponential and faster during the first hour possibly due to the presence of molecules of the compound on the surfaces of the particle. After 2 hours the released was delayed possibly due to the diffusion of C6I dissolved in the nucleus of the nanoparticles in the dissolution medium.
In this work, the release profile showed that C6I improved its solubility in solution when it was incorporated into Precirol® NPs compared to the results of the intrinsic dissolution of pure free C6I. According with the IDR value, 0.24 mg of compounds per centimeter was dissolved each minute, while the release and dissolution of C6I in the nanoparticles occurred rapidly in the first hours and more than half of the compound was in solution. According to Noyes-Whitney and Ostwald-Freundlich principles, the particle size in the nanometer range can lead to an increased dissolution velocity and saturation solubility for a nanoformulation. Previous studies with a number of poorly soluble drugs have demonstrated that particle size reduction can lead to an increased rate of dissolution and higher oral bioavailability [8,29,36].
On the side of toxicity and effectiveness, our results showed a significant decrease in the toxicity but an increase of antileishmanial activity for intracellular amastigotes of L. (V) braziliensis of C6I when incorporated in Precirol® NPs. In this case, the EC50 of PC6I was 40-fold lower than free C6I. Additionally, previous studies in vivo demonstrated that C6I was able to cure hamsters with CL by L. (V) panamensis when administered topically at a dose of 2 mg/kg/day for 15 days [5]. Here we show that free C6I, administered orally at a dose of 15 mg/kg/day for 28 days, showed an effectiveness close to 30%. Nevertheless, its effectiveness increased to 71% when the C6I was incorporated in Precirol® NPs and administered at a higher dose of 30 mg/kg/day. The results were consistent in the in vivo model, with 71% cure in the group of animals treated with PC6I upon oral administration of a dose of 30 mg/kg/day for 28 days. Differences in the effectivity and toxicity between free C6I or encapsulated PC6I was evident not only because the LC50 increased two-fold but also because the EC50 was lowered 40-fold. Several hypotheses may explain how the SLN improve de effectivity of compound. We hypothesize that the solubility and dissolution of pure compound was low so that the concentration available in polar and physiological solutions is few. Possibly, nanoparticles loaded with C6I increased the dissolution of compound and improved the solubility in polar solutions, for example, culture medium, and thus, more compound was available to enter in the infected cell. In the case in vivo test it is possible that compound was soluble in GI solutions and thus, the dissolved C6I is able to cross the intestinal wall and to reach the systemic circulation and its target [8,14,29,37]. Furthermore, SLNs used for oral administration offer several benefits over conventional formulations including increased solubility, enhanced stability, improved epithelium permeability and bioavailability, prolonged half-life, tissue targeting, and minimal side effects as well as the potentiating effect of the antileishmanial activity [8,28].
The in vivo activity of free C6I or incorporated in the Precirol® NPs (PC6I) was tested in the hamster because this is the most adequate animal model for CL by Leishmania species of Viannia subgenus. The effectiveness was compared with the intramuscular MA instead miltefosine because MA is able to resolve CL in the hamster model infected with L. (V) braziliensis at the dose used in this study [20] while miltefosine has not shown any cure [38]. Despite the use of different administration routes, both treatment with the salts and the MA are systemic and this validates the use of MA as a control. On the other hand, although the days of treatment were different (28 vs. 10 for the salts vs. MA), the efficacy of the treatments was evaluated based on the outcome of the cure observed after the treatment was finished. In this way, day 0 of follow-up began one day after the end of the treatment, regardless of the duration of the treatment. Moreover, the treatment for all groups was safe during 28 days of administration and during 90 days of follow-up; the hamsters gained weight and levels of ALT, BUN, and CRT were within the range of normal values for all animals both before (TD0) and after (PTD8) treatment.
Additionally, in all assays where the safety profile was tested, the compound was safe. This profile was studied in rats and mice as recommended in the international guides; in this way it is also guaranteed that the non-toxicity of the substance does not depend on the species tested. We also showed that this activity is selective, in vitro being potentially non-toxic for colon Caco-2 cells and for primary cultures of human macrophages huMDM, which suggest that C6I does not affect the colon cells during the process of intestinal absorption nor the host cells for Leishmania parasites during treatment. Moreover, the NPs improved the cytotoxicity of the free C6I increasing the LC50 in 21 times for the U937 and MPha. The LC50 for Caco-2 and huMDM were similar. The in vivo oral acute toxicity results defined the maximum concentration to evaluate the effect after oral administration at repeated doses (28 days). There were no deaths or changes in body weight, as well as anatomical changes at necropsy that could be associated with a possible toxicity for C6I. The PC6I formulation presented a pattern of safety as shown by results obtained in organs such as heart, spleen, intestine, brain and reproductive organs, as well as the blood chemistry profile without evident toxic manifestations. Although, two animals had fatty liver and at the histological level, events such as congestion and mild to moderate vacuolar degeneration were observed, it is known that NP systems induce oxidative stress in the liver [39]. Additionally, the presence of a cationic surfactant may generate some cytotoxicity [40].
In conclusion, despite the low solubility and the dissolution profile shown by C6I, the compound possesses other physicochemical, pharmacokinetic and pharmaceutical properties that make it suitable for the oral formulation. The C6I showed good intestinal permeability and potential no toxicity after oral administration in the murine model. The genotoxicity or mutagenicity of C6I were absent. The Precirol® NPs was a suitable strategy that improved not only the bioavailability and toxicity but also the effectiveness of the C6I both in vitro and in vivo. Indeed, the cure rate of hamsters with experimental CL increased from 30% to 71% after daily administration for up to 28 days. Results showed here confirm that the PC6I formulation has high potential to become a new drug for the treatment of CL.
MF, TG, AA, SMR carried out the experiments. The manuscript was prepared by MF, SMR. All authors analyzed the data, discussed the results and revised the manuscript.
The authors declare that there is no conflict of interest in this work.
This work was supported by The Colombian Department of Science, Technology and Innovation-Colciencias [CT-695-2014]; M.F.C had a financial support of Colciencias (CT 567-2014).
To D. Gaspar, A. Restrepo, N. Arbelaez and D. Garcia for technical assistance.