Valosin Containing Protein (VCP) gene mutations have been reported in ~1-2% of familial amyotrophic lateral sclerosis (ALS). We report a case of clinically defined and neuropathologically confirmed ALS in a 48-year-old, emaciated female with a p.R155C (c.463 C > T) mutation in VCP gene. She presented with progressive generalized muscular weakness, weight loss, dyspnea on exertion, combined nasal and spastic dysarthria, positive jaw jerk and exaggerated gag reflex. Electrodiagnostic studies revealed involvement of both upper and lower motor neuron typical of generalized ALS. She died of fulminant ALS three years after onset of clinical features. Neuropathological examination revealed extensive spinal motor neuron degeneration with loss of spinal anterior horn cells, gliosis and mislocalization of TDP-43 positive inclusions. Bilateral lateral columns showed degeneration. No abnormalities were detected in the brainstem and cerebellar hemispheres except for occasional intraneuronal vacuolation in the medullary nuclei and Bunina bodies in the hypoglossal nucleus.
Valosin containing protein, IBMPFD, Amyotrophic Lateral Sclerosis, TDP-43, vacuolation
ALS: Amyotrophic Lateral Sclerosis; AchR: Acetylcholine Receptor; EMG: Electromyography; FUS: Fused in Sarcoma/Translated in Liposarcoma; FTD: Frontotemporal Dementia; IBMPFD: Inclusion Body Myopathy, Paget's Disease of Bone and Frontotemporal Dementia; IBMFRS: Inclusion Body Myopathy Functional Rating Scale; MRI: Magnetic Resonance Imaging; MuSK: Muscle Specific Kinase; PDB: Paget's Disease of Bone; SNIP: Spontaneous Nasal Inspiratory Pressure; SOD1: Cu/Zn-Superoxide Dismutase; TDP 43: TAR DNA-Binding Protein 43; VCP: Valosin Containing Protein
Inclusion Body Myopathy associated with Paget's disease of bone and Frontotemporal Dementia (IBMPFD OMIM#167320) [1]) or more recently described as multisystem proteinopathy [2] is an autosomal dominant disorder caused by mutations in the Valosin Containing Protein (VCP) gene first identified by Watts, et al. [3,4]. Approximately 90% of the mutation positive individuals develop myopathy in their 30s-40s manifesting as shoulder and pelvic girdle muscle weakness and atrophy; later progressively involving other muscle groups; 50% develop Paget disease of bone (PDB) typically in their 30s and 30% develop frontotemporal dementia (FTD) in their mid-50s [3,5] in which the characteristic changes in personality and progressive loss of language precede memory loss. Individuals typically die from cardiac or respiratory failure in their 40s-60s [3,5]. Mutations in VCP gene have also been associated with non-syndromic familial FTD [6] and 1-2% of familial ALS [7]. ALS typically presents in 20s and 60s [8]. Other phenotypic features of VCP disease have been reported including Parkinson's disease, cardiomyopathy, hepatic steatosis, sensory-motor axonal neuropathy, and sphincter disturbance. The diagnosis is typically made based on the presence of proximal myopathy, rimmed vacuoles, ubiquitin and TDP-43 positive inclusions in the affected tissues and typically the co-existence of PDB, FTD and/or ALS [3,5,9-11]. Here, we report clinicopathological findings in a patient with VCP gene mutation p.R155C (c.463 C > T) who died approximately three months after the clinical diagnosis of generalized ALS.
This 48-year-old emaciated woman was evaluated for marked, rapidly progressive muscle weakness with widespread fasciculations and dysarthria. She had been previously healthy and an athlete throughout her life. She had a positive family history of myopathy and PDB in her father, brother, daughter and multiple other family members (Figure 1). Three other relatives in the same and in a previous generation were also found to have possible features of a denervating disorder although the data is insufficient to allow any certainty as to whether they also had ALS like phenotype. Her past medical history was significant for malignant thymoma with metastasis to both kidneys and lungs diagnosed at age 30 years. She required chemotherapy and surgery and had been in full remission since age 31 years. There were no associated clinical or laboratory findings of myasthenia gravis. At the age of 45 years she developed progressive muscle weakness in her lower back and lower extremities and rapidly deteriorated over the subsequent three years to develop quadriparesis of her extremities, became wheelchair-bound and was unable to perform activities of daily living. She developed dysphagia for both liquids and solids and her weight dropping from 140 pounds to 117 pounds. She had no cognitive and behavioral dysfunction; however, was severely depressed without suicidal ideations. She received treatment for chronic neck and back pain but had no evidence of Paget's disease of bone.
 Figure 1: Pedigree of our patient with familial myopathy, ALS, Paget's disease of bone and dementia.
View Figure 1
Figure 1: Pedigree of our patient with familial myopathy, ALS, Paget's disease of bone and dementia.
View Figure 1
On neurological examination she has marked tetraparesis, with weakness and atrophy of her both upper and lower limb girdle muscles; tongue and facial muscles and neck flexors. There was also spasticity in lower limbs with widespread fasciculations of the limbs and tongue, a positive jaw jerk and exaggerated gag reflex. She had severe bulbar dysfunction with severe spastic dysarthria difficulty swallowing and drooling. Sensory function was normal, and coordination could not be tested. She was severely dyspneic and orthopneic. Bladder and bowel function were normal.
Functional assessment using IBM functional rating scale (IBMFRS) [12] demonstrated a very low score of 3 (range: 0-40). The biochemical and immunological work up including serum creatinine phosphokinase, alkaline phosphatase, acetylcholine receptor (AChR) binding antibodies and muscle specific tyrosine kinase (MuSK) antibodies for myasthenia gravis were negative. Post-contrast spine Magnetic Resonance Imaging revealed severe atrophy and fatty replacement of the paraspinal muscles. She had severe restrictive pattern respiratory insufficiency with a predicted forced vital capacity in erect posture of 13% (0.46 L) and a sniff nasal inspiratory pressure (SNIP) of -11 cm of H20 suggestive of severe diaphragmatic insufficiency. Nerve conduction studies showed normal sensory responses and reduced compound motor action potentials in all motor nerves (median, ulnar, peroneal and tibial). Needle electromyography (EMG) of the muscles of the right arm, right leg, C5-T1, L2-S1, and right sternomastoid muscles showed fibrillation potentials in all the muscles. Motor units showed large amplitude, broad duration, polyphasic potentials with marked reduction in recruitment and interference patterns and very prominent fast firing. No myopathic potentials were detected. The clinical presentation and progression, widespread neurogenic changes including acute and chronic, with 3 to 4+ fibrillations and fasciculation on needle EMG suggestive of reinnervation and chronic denervation with unit remodeling in three separate regions of the body was diagnostic of ALS. She died approximately three months after the clinical diagnosis of generalized ALS. Her husband consented to an autopsy. We report the clinicopathological findings which confirmed the clinical findings of ALS.
An autopsy of the nervous system revealed a brain weight of 1316 grams post fixation. Gross examination of the cerebral and cerebellar hemispheres revealed mild generalized atrophy with well-preserved deep gray matter structures.
Microscopically, cerebral cortical sections showed mild cortical atrophy with mild subpial gliosis, rare rimmed eosinophilic inclusions in some thalamic neurons and neuronal cytoplasmic clearing around centrally placed nuclei. Occasional ubiquitin positive nuclei in cortical neurons were observed. No specific abnormalities were identified in other brainstem nuclei and cerebellar hemispheres except for occasional intraneuronal vacuolation in the medulla and eosinophilic inclusions (Bunina bodies) in the neuronal cytoplasm of the hypoglossal nucleus and Purkinje cell layer of cerebellum.
Significant pathology of the spinal cord was noted, however. There was anterior horn cell loss with gliosis and macrophagic infiltration involving the lateral and anterior funiculi of the spinal cord. Lipid laden macrophages indicated on-going degeneration of the lateral columns (Figure 2). Mislocalization of TDP-43 positive inclusions were seen in numerous motor neurons with loss of nuclear staining and scattered positive inclusions in the neuropil of the anterior horns.
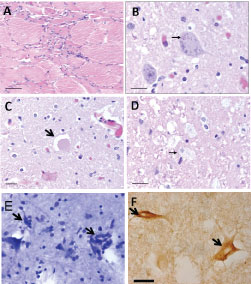 Figure 2: Histological analysis of skeletal muscle, spinal cord and cerebrum. a) Examination of the quadriceps muscle revealed grouped atrophy of quadriceps muscle, indicating chronic neurogenic disease (scale bar = 50 μm); b) There were Bunina bodies (arrow) within the cytoplasm of an anterior horn lower motor neuron (scale bar = 20 μm) and scattered hyaline eosinophilic inclusions in the anterior horns of the spinal cord and occasional glassy cytoplasm in the motoneurons; c) Axonal swelling (arrow) involving the proximal axon segment in the spinal cord anterior horn (scale bar = 20 μm); d) Lipid laden macrophages (arrow) indicated on-going degeneration of the lateral columns (scale bar = 20 μm); e) Toluidine blue stain of primary motor cortex showed neuronal loss, gliosis, and microglial activation with neuronophagia (arrows); f) TDP-43 immunostain showed cytoplasmic staining in numerous motor neurons of the anterior horns.
View Figure 2
Figure 2: Histological analysis of skeletal muscle, spinal cord and cerebrum. a) Examination of the quadriceps muscle revealed grouped atrophy of quadriceps muscle, indicating chronic neurogenic disease (scale bar = 50 μm); b) There were Bunina bodies (arrow) within the cytoplasm of an anterior horn lower motor neuron (scale bar = 20 μm) and scattered hyaline eosinophilic inclusions in the anterior horns of the spinal cord and occasional glassy cytoplasm in the motoneurons; c) Axonal swelling (arrow) involving the proximal axon segment in the spinal cord anterior horn (scale bar = 20 μm); d) Lipid laden macrophages (arrow) indicated on-going degeneration of the lateral columns (scale bar = 20 μm); e) Toluidine blue stain of primary motor cortex showed neuronal loss, gliosis, and microglial activation with neuronophagia (arrows); f) TDP-43 immunostain showed cytoplasmic staining in numerous motor neurons of the anterior horns.
View Figure 2
Microscopic examination of muscle revealed group atrophy, with scattered plump, enlarged myofibers and numerous internally located nuclei. There were rows and groups of nuclei and the presence of concentric eosinophilic inclusions in the cytoplasm of some myocytes. Positive ubiquitin nuclear staining with some subsarcolemmal accumulation and round ubiquitin+ inclusions were observed. The pathologic findings were more pronounced in the biceps sections than in the quadriceps. Immunostains for tau were universally negative.
This case is illustrative of a rare aspect of VCP disease- fulminant classic ALS associated with upper and lower motor neuron signs, progressive generalized muscular weakness, spasticity, weight loss, dyspnea, spastic dysarthria, positive jaw jerk, exaggerated gag reflex, and neuropathological findings of extensive spinal motor neuron degeneration with loss of spinal anterior horn cells, gliosis and mislocalization of TDP-43 positive inclusions.
ALS is a progressive, lethal neurodegenerative disease with loss of motor neurons in spinal cord, cerebral cortex and brain stem. Mutations in several genes have been identified in familial ALS including mutations in TDP-43 [13,14], FUS [15], optineurin [16], VCP [17] and most recently C9ORF72 [18]. VCP has also been recently associated in the etiology of sporadic ALS [17,19,20]. To date over 40 coding mutations have been identified in the VCP gene [7,8,19,21-23]. Although Tucker, et al. in 1984 [24] reported a family with typical combined lower motor neuron degeneration, and skeletal disorganization, the clinical spectrum associated with VCP mutations has recently been extended to include ALS [16,25]. Using whole-exome sequencing Johnson, et al. identified a p.R191Q mutation in VCP in a family with autosomal dominant ALS [7]. Screening of additional 210 familial and 78 ALS patients confirmed by autopsy revealed four additional individuals with VCP missense mutation (1-2% of familial ALS). The Tucker family were later found to have a p.R155P VCP mutation by Watts, et al. in 2004 [4].
VCP is a ubiquitously expressed protein belonging to the type II AAA ATPase superfamily. VCP is involved in many cellular functions, the most important being related to protein degradation by the ubiquitin proteasome and autophagy pathway in which VCP helps deliver ubiquitinated substrates to the 26S proteasome for degradation [26]. VCP mutations are also reported to cause mitochondrial dysfunction and hence reduced ATP production [27]. Buchan, et al. reported the role of VCP in the clearance of stress granules that are accumulated as TDP43 in the affected individuals with VCP mutations [28]. TDP-43 and ubiquitinated proteins are the major component of inclusions characteristic of VCP-associated pathology, placing VCP disease in a novel category of neurodegenerative diseases termed TDP-43 proteinopathies [4,22]. Among the previously reported families with VCP mutations R159G, R159C, R159H, 191G, R191Q or D592N, ALS was the predominant phenotype and patients with I114V and D592N presented with pure ALS [8]. VCP gene mutations has been reported in families presenting with myopathy and/or PDB, FTD and other less common phenotypes [22]. Given the shared pathogenesis with other neurodegenerative diseases Benatar, et al. have labelled this group of disorders as multisystem proteinopathies [2].
We recently produced a knock in mouse harboring the most common VCP mutation associated with human disease (R155H) [29,30] and in a subsequent study examined and characterized for the first time the evolution of progressive ALS-like pathological changes in the spinal cords of these animals, establishing this VCP mouse as a new ALS related animal model [31] that may prove to be of value for studying disease mechanisms and assessing therapeutics.
This case provides a unique opportunity to study the clinical and pathologic features of generalized ALS emphasizing on astute physical examination and the value of electrodiagnostic studies in the final diagnosis. Individuals with VCP disease exhibiting a rapid rate of deterioration should be specifically evaluated for ALS by clinical and EMG studies. VCP molecular testing has recently been added to the panel of genes currently employed for testing individuals with familial ALS.
We thank the patient and her family for permission to publish these studies, the MitoMed Laboratory for molecular testing, and the Institute for Clinical and Translational Science (ICTS), University of California-Irvine for the resources for the clinical studies.
No conflicts of interest.