Alzheimer's disease (AD), the most common type of dementia or neurodegenerative diseases since the beginning of the past century when it was coined by the German psychiatrist, Alois Alzheimer in 1901 holds an allusive etiology, and as a result failed most treatment and prevention strategies. Despite the common belief among the experts of AD being a genetic disease, the genetic heritability of this disease has such a wide range of 50-80% in twin and family studies that questions the totality of it as being hereditary. To understand the etiology or in a better word the Pathophysiology of Alzheimer's disease even in its genetic format, the science needs to dig beyond the genetics that's only a pathway of faulty gene mutations transcriptions and transmissions. In relation to AD, in fact as early as 1980's, there has been report and scientific suspicion as viral insults being a cause of the disease. While in the past there was disbelief about viral existence in our brains, Itzhak in 1994 reports of such finding in his lab, that herpes simplex type I virus (HSV1) DNA, the cause of common and recurrent oral herpes or "cold sores", is present in many normal aged brains and AD brains, but absent in brains of younger people. The viral association with AD, Specially HSV1 has been reported as early as 1980 by Middleton and colleagues.
In this paper through a selective review of the literature, specifically on the Pathophysiology of Alzheimer's disease, we have tried to show the common epigenetic risk factors that lead the brain to Neurodegeneration in genetic and non-genetic cases of the disease.
Alzheimer's disease (AD) which is the most common type of dementia and accounts for up to 70% of such cases, carries its label from the German psychiatrist, Alois Alzheimer [1] who identified the first case of such condition in a 50-year-old woman in 1901. Despite the first reported case by Dr. Alzheimer's being young, the early onset AD is rare and the majority of cases are first diagnosed after age 65 and the risk of disease is higher with the older age, so that incidence rises from 3% in 60's to about 25-40% in 80's and about 70% in 90's [2]. In fact and as we will read further here, senescence and the old age is associated with some cognitive and memory loss due to the aging of the brain and its neurons like aging of the rest of the body and loss of its functions and youth.
Despite the common belief among the experts of AD being a genetic disease, the genetic heritability of this disease has such a wide range of 50-80% in twin and family studies that questions the totality of it as being hereditary. Moreover genetics or mutations of genes are not causes of disorders but paths of transcriptions and transmissions. Therefore the common sense dictates to search for a cause(s) that lead(s) to such faulty mutations, then transcriptions and transmissions so fooling us to perceive genetics as cause(s) of diseases! In relation to AD, in fact as early as 1980's, there has been report and scientific suspicion as viral insults being a cause of the disease. While in the past there was a disbelief about viral existence in our brains, Itzhak in 1994 [3] reports of such finding in his lab, that herpes simplex type I virus (HSV1) DNA, the cause of common and recurrent oral herpes or "cold sores", is present in many normal aged brains and AD brains, but absent in brains of younger people. The viral association with AD, Specially HSV1 has been reported as early as 1980 by Middleton and colleagues [4].
The herpes virus, both HSV1 and HSV2 (responsible for genital herpes) are both neurotropic and neuro-invasive, meaning they persist in the body by becoming latent and hiding from the immune system in the cell bodies of neurons. After the initial or primary infection, the virus in a nerve cell becomes active and is transported via the neuron's axon to the skin, where virus replication and shedding occur and cause new sores. HSVs may persist in a quiescent but persistent form known as latent infection, notably in neural ganglia, e.g. HSV-1 tends to reside in the trigeminal ganglia, while HSV-2 tends to reside in the sacra ganglia and express "latency Associated Transcript (LTA) RNA. LAT is known to regulate the host cell genome and interferes with natural cell death mechanisms. By maintaining the host cells, LAT expression preserves a reservoir of the virus, which allows subsequent, usually symptomatic, periodic recurrences or "outbreaks" characteristic of non-latency. Whether or not recurrences are noticeable (symptomatic), viral shedding occurs to produce further infections in the victim or a new host, through contact [5-7].
In the presence of a certain gene variation (APOE-epsilon4 allele carriers), there is more chance of developing AD in the presence of HSV-1 and aged or inactive brain neurons. The mechanism that the virus degenerates the brain cells and causing AD is by recruiting cell membranes from the host containing Amyloid precursor protein (APP), and consequently leads to deposition of Amyloid plaques in the brain, replacing neurons [8]. According to some studies, without the presence of the above-mentioned gene allele, HSV-1 and aged or inactive brain, such damaging or neurodegenerative process would not occur in the brain and there is low or no risk of developing Alzheimer's. A recent prospective study published in 2008 with a cohort of 591 subjects [7] showed a statistically significant difference between patients with antibodies indicating recent reactivation of HSV and those without these antibodies in the incidence of Alzheimer's disease, without direct correlation to the APOE-epsilon4 allele. In 2011 Manchester University scientists [9] showed that treating HSV1-infected cells with antiviral agents decreased the accumulation of β-amyloid and P-tau, and also decreased HSV-1 replication.
While there is a strong link between HSV1 and the development of AD, it seems that the herpes simplex virus would not lead to the development of Alzheimer's in every brain that it invades [10]. A recent study by Wozniak research team in 2009 [11] have showed that while in Alzheimer's disease brains, 90% of the plaques contained the viral DNA and 72% of the DNA was associated with plaques, the control aged normal brains also contained amyloidal plaques, though at a lower frequency, and 80% of plaques contained herpes simplex virus type 1 DNA, though only 24% of the viral DNA was plaque-associated. These researchers have suggested that "this is because in aged normal individuals, there is a lesser production and/or greater removal of beta-amyloidal (Abeta), so that less of the viral DNA is seen to be associated with Abeta in the brain." Therefore as it was stressed earlier, HSV1 can only lead to AD in the presence of APOE-4 gene plus aged or inactive brain [12,13].
The complex mechanism that HSV1 or any other possible virus invades the human body and the brain is worth detailing. Viruses can be conceptualized as self-replicating multi-protein assemblies, containing coding nucleic acids. Viruses have evolved to exploit host cellular components including enzymes to ensure their replicative life cycle. New findings indicate that also "Viral Capsid Proteins" recruit host factors to accelerate their assembly. These assembly machines are RNA-containing multi-protein complexes whose composition is governed by Allosteric sites. In the event of viral infection, the assembly machines are recruited to support the virus over the host and are modified to achieve that goal. In the case of viral infection, the assembly machines have been modified by the virus to meet the virus' need for rapid Capsid assembly rather than host homeostasis. In the case of the neurodegenerative diseases, it is the monomers and/or low n Oligomers of the so-called aggregated proteins that are substrates of assembly machines. Examples for substrates are Amyloid peptide and tau in Alzheimer's disease, synuclein in Parkinson's disease, Prions in the Prion diseases, and others. The diagram below show how the viral and "viral Capsid proteins" form these assemblies in favor of survival of the viral machinery in the host body such as human's [14] (Figure 1).
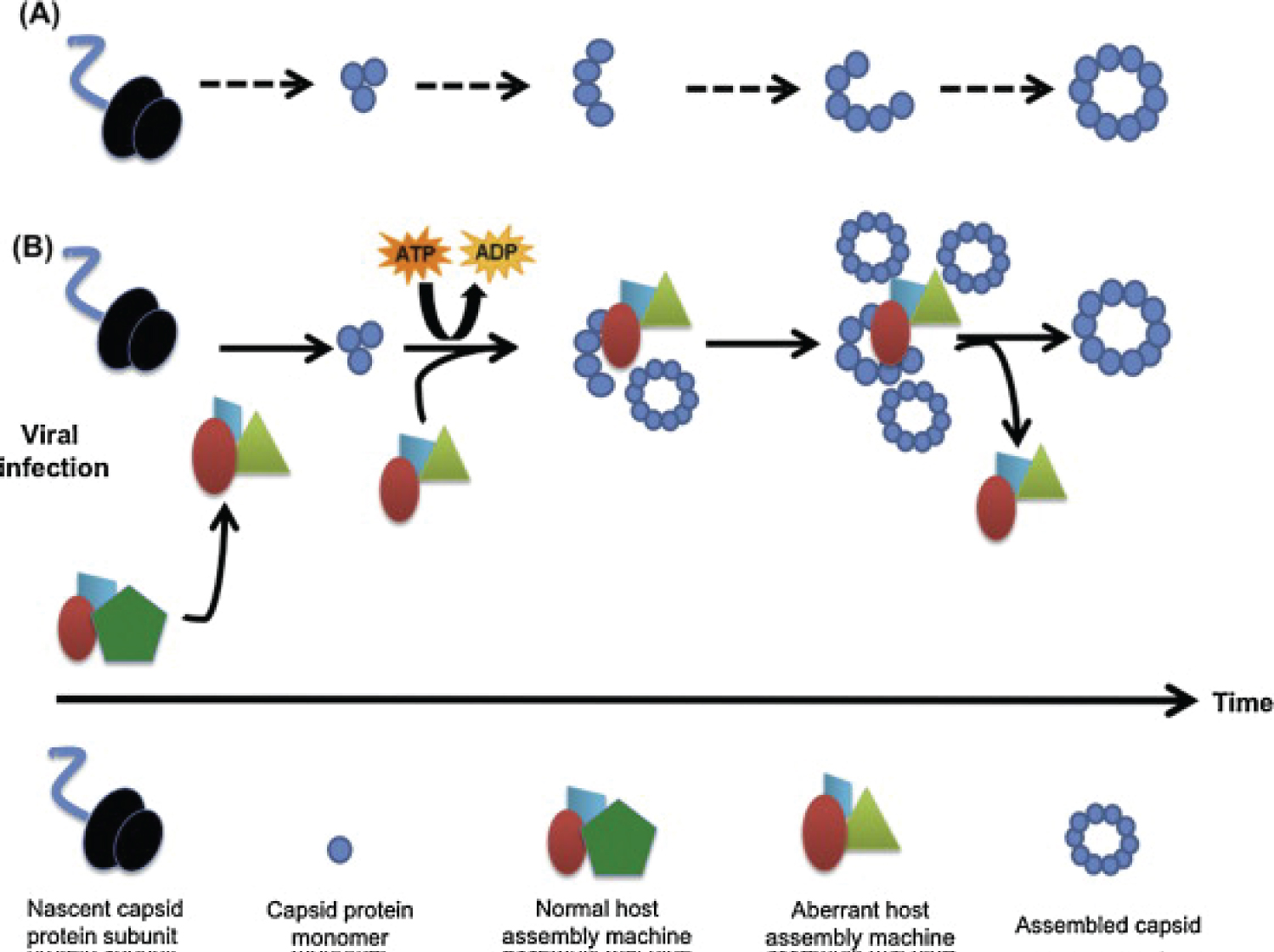 Figure 1: The figure shows how the viral and "viral capsid proteins" form these assemblies in favor of survival of the viral machinery in the host body such as human's.
View Figure 1
Figure 1: The figure shows how the viral and "viral capsid proteins" form these assemblies in favor of survival of the viral machinery in the host body such as human's.
View Figure 1
Recently, findings comprising various viral families suggest that the cellular pathway to Capsid assembly may be substantially different from the foundational self-assembly models that conform to the minimum requirements of thermodynamics. Indeed, without such catalytic "add ons" viral propagation might not be possible, given the obstacles not only of cytoplasmic crowding but also of innate immune mechanisms. Specifically, it has been suggested that Capsid assembly is accelerated by transient formation of virus-recruited multi-protein complexes with enzymatic activity that serve as "assembly machines" to accelerate Capsid formation. This catalyzed viral Capsid assembly is an energy-dependent process, and host protein factors seem to play a major role in this. In summary, surprising similarities in the cellular biology of virus Capsid assembly and endogenous protein assembly suggest that cellular host factors, i.e. assembly machines, assist in and accelerate protein multimerization. Through their rapid generation cycles compared to their host cells, viruses have identified and exploited cell-resident macromolecules provided by host proteins used them to their advantage, i.e. fast and stable virus replication including Capsid assembly [15].
Alzheimer's Disease's pathology that is well known to be due to deposition of Amyloid plaques in the brain, hence replacing healthy normal neurons and causing Neuro-degeneration, all happen y a microbial invasion of a virus such as Herpes long ago during the young ages of the victims. The mechanism that the virus Degenerates the brain cells and causing AD is by recruiting cell membranes from the host containing Amyloid precursor protein (APP), and consequently leads to deposition of Amyloid plaques in the brain. But the virus needs a stale, aged or inactive brain, to cause such damage. Treating HSV1-infected cells with antiviral agents could decrease the accumulation of β-Amyloid and P-tau, and also lower the viral replication in the aged brain. But an active brain even if aged and infected with HSV in youth has a chance of lesser production and/or greater removal of beta-Amyloid (Abeta) to degenerate the brain cells and cause dementia or Alzheimer's. In a detailed Pathophysiology word, the viral assembly machine cannot use or recruit the active healthy neurons of the host at its service to cause damage and Neuro-degeneration.