Background: Interstitial lung disease (ILD) is one of the most serious complications among patients with scleroderma. It is associated with significant morbidity and mortality. Little is known about the epidemiology of scleroderma associated ILD in sub-Saharan Africa. Thus, we aimed to determine the prevalence, clinical characteristics and outcomes of patients with scleroderma -ILD.
Methods: A retrospective electronic chart review was conducted of patients with systemic sclerosis seen between January 2010 and December 2016 in the Departments of Pulmonology and Rheumatology at Inkosi Albert Luthuli Central Hospital, Durban, South Africa.
Results: A total of 146 patients with systemic sclerosis (SSc) were seen during the study period. Fifty-five had SSc-associated ILD, giving a prevalence of 37.6%. The median age was 51 (IQR 41-60) years, 87% of patients were female and 56% were of Indian descent.
Dyspnea was the presenting complaint in 47 (85.4%) patients, while 16 (29.1%) presented with cough. ANA was positive in 50 patients (90.9%), anti-Scl-70 antibodies were positive in 21 (38.2%). Thirty-seven patients (67.3%) received immunosuppressants, with 27 (49.1%) receiving cyclophosphamide as induction therapy, while 18 (32.7%) patients did not receive any specific therapy. Most patient symptoms remained static during the period of observation. UIP was the most common radiological diagnosis. Follow-up CT scans were available in 48 (87%) patients, with the majority of patients showing no significant radiological changes between their first and last CT scans. Follow-up lung function testing showed a statistically significant decrease in median FVC of 0.09l (p = 0.011). Overall 20 (36.4%) patients had a significant decline in FVC, while 7 (12.7%) had an improvement in FVC.
Conclusion: ILD is common in systemic sclerosis, affecting 1 in 3 patients with systemic sclerosis in our cohort. Immunosuppressant treatment may arrest or retard the rate of decline of lung function.
Scleroderma, Interstitial lung disease, KwaZulu-Natal-South Africa
Systemic sclerosis (SSc) is a systemic autoimmune disorder characterized by excessive extracellular matrix production, due to a small vessel vasculopathy coupled with immune dysregulation. The production of autoantibodies causes fibroblast dysfunction and results in fibrosis of the skin and internal organs [1]. The pathogenesis of SSc remains unclear, even though recent research has focused on a variety of pathways that might lead to new therapeutic targets [2].
Pulmonary involvement is common in patients with SSc and most often comprises fibrosis or interstitial lung disease (ILD), and pulmonary vascular disease leading to pulmonary arterial hypertension (PAH). Using pulmonary function tests (PFTs), significant pulmonary involvement is detectable in 25% of patients with SSc within 3 years of initial diagnosis [3]. Pulmonary manifestations are the leading cause of disease-related morbidity and mortality in patients with SSc. A recent report estimated the mortality from pulmonary disease in SSc patients to be 33% [4]. Interstitial lung disease is more common among African Americans and in people with the diffuse cutaneous form of systemic sclerosis or anti-topoisomerase 1 antibodies. Systemic sclerosis-associated interstitial lung disease most commonly presents with dyspnoea, cough, and a non-specific interstitial pneumonia pattern on CT scan, with a minority of cases fulfilling the criteria for usual interstitial pneumonia.
The standard therapy has traditionally been combinations of immunosuppressants, particularly mycophenolate mofetil or cyclophosphamide [5,6]. Glucocorticoids are still frequently used but have shown limited benefit and may increase risk of scleroderma renal crises (10.12). Although definitive data are lacking, mycophenolate mofetil (MMF) appears to improve patient outcome in SSc-ILD with a reasonable safety profile, and may be considered as first line therapy in these patients [7-11]. Azathioprine (AZA) has not been well investigated but appears to be a well-tolerated agent for maintenance therapy in patient with SSc-ILD [7,12,13].
There is limited data available on scleroderma-associated interstitial lung disease in Southern Africa. A retrospective review of SSc patients attending a tertiary connective tissue diseases clinic compared patients with and without ILD [14].
Given the probable role of genetic and/or environmental factors in the pathogenesis of this disease, and in the response to therapy, it is important to evaluate differences in geographic regions and patient populations. The aims of the study were: a) To determine the patient profile and b) Outcomes of patients with scleroderma-associated interstitial lung disease in Durban, KwaZulu-Natal, South Arica.
This study was a retrospective observational descriptive study using data obtained from preexisting electronic patient records. It was conducted at Inkosi Albert Luthuli Central Hospital, an 800-bedded central referral hospital in Durban, KwaZulu-Natal South Africa. The study included all adult patients (18 years and older) diagnosed with scleroderma with interstitial lung disease who were seen in the pulmonology clinic between 01/01/2010 and 31/12/2016. The clinic is a referral clinic accepting patients throughout the province of KwaZulu-Natal. The study excluded patients where electronic data sets could not be retrieved due to technical issues, those younger than 18 years and patients with other connective tissue diseases.
Demographic and clinical data, as well as data from special investigations, were captured retrospectively from medical records. Demographic data included age, gender and race. Clinical data captured were presenting symptoms, pre-existing medical conditions functional class, date of onset of initial symptoms, and treatment. Special investigations captured included results of HRCT, antinuclear factor (ANA), anti-Scleroderma-70 (Scl-70), and anti-centromere antibody (ACA) levels, and PFT. There is no valid definition of SSc-ILD disease progression, but we defined a decline of ≥ 10% in any pulmonary function parameter as being significant deterioration, with an increase of at least ≥ 10% from base line as being significant improvement.
Ethical approval was granted by the University of KwaZulu-Natal's Human Research Ethics Committee (ref. no. BE602/18) and the KwaZulu-Natal department of Health (ref. no. KZ_201905_016).
Statistical analysis was performed using SPSS version 27. Basic descriptive statistics were performed with continuous variables expressed as median and interquartile range (IQR) and categorical variables expressed as number and proportions. Categorical variables were compared using the Chi-square test or Fisher's exact test as appropriate. The related-samples Wilcoxon signed-rank test was used to compare first and last pulmonary function test results. A p-value of < 0.05 was deemed statistically significant.
A total of 146 patients with systemic sclerosis were seen during the study period. Fifty-five of these patients had SSc-associated ILD. The prevalence of ILD in systemic sclerosis was 37.6%.
In the patients with SSc-ILD, the median age was 51 years (IQR 41-60). Forty-eight (87%) were female and 31 (56%) were Asian with 22 (40%) being black Africans. Forty-one had other co-morbidities, including overlap with other connective tissue diseases (Table 1). Eighteen (32.7%) patients had pulmonary hypertension, the median pulmonary arterial pressure on echocardiography was 43 mmhg (IQR 40-60) and the maximum recorded PAP was 100 mmhg. Four (7.3%) patients had osteoporosis. Three patients were HIV positive. All 3 were on antiretroviral therapy and their CD4 counts ranged from 568 to 630 cells/ul.
Table 1: Baseline demographic data. View Table 1
Details of patient's presenting symptoms are shown in Table 2 and a summary of non-pulmonary organ involvement is shown in Figure 1.
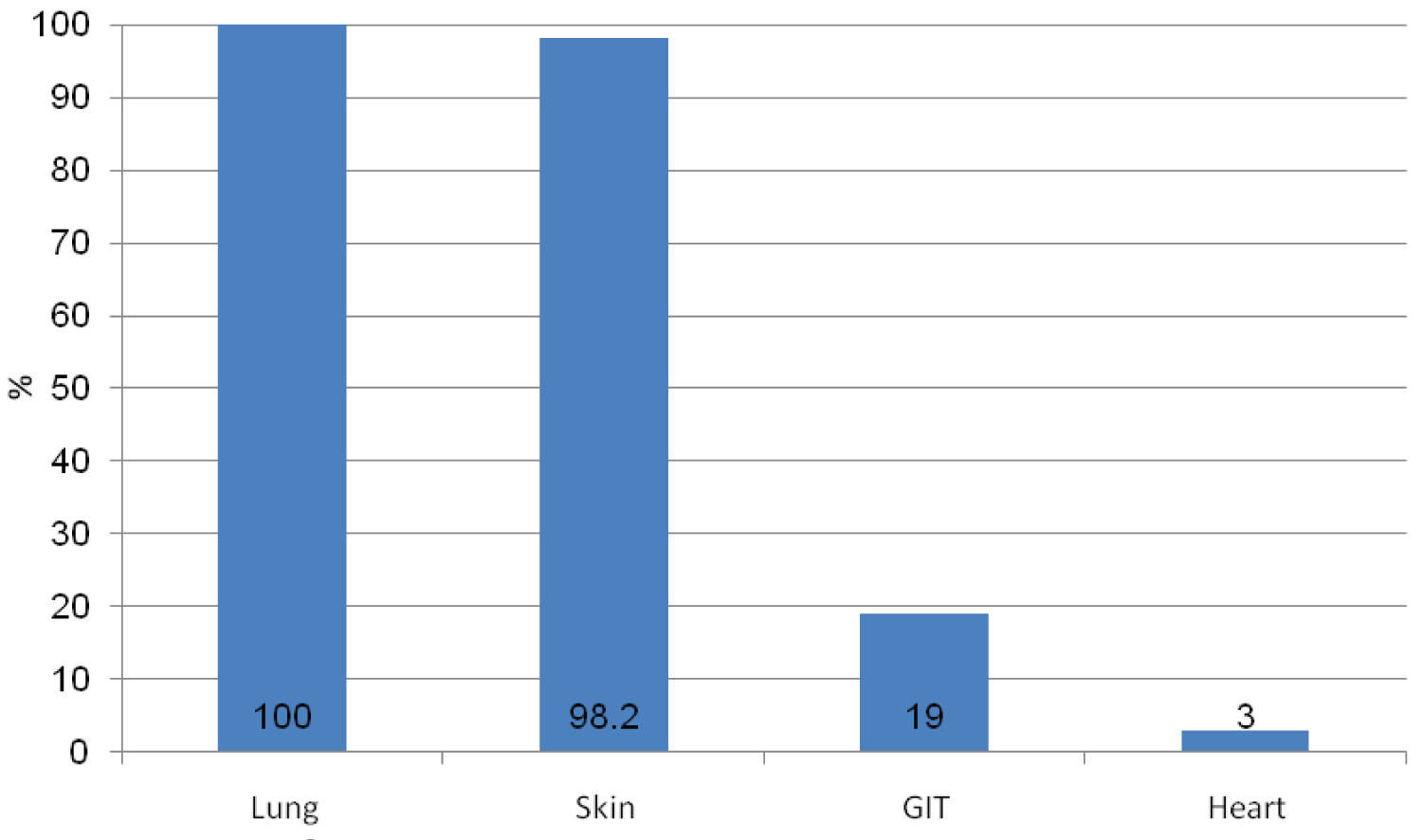 Figure 1: Non-pulmonary organ involvement in patients with SSc-ILD.
View Figure 1
Figure 1: Non-pulmonary organ involvement in patients with SSc-ILD.
View Figure 1
Table 2: Presenting symptoms. View Table 2
The results of serological testing are shown in Table 3. The median ANA titre was 800 (IQR 200-2560), with the median anti-Scl-70 titre being 141 (IQR 38-320) anti-centromere antibodies were only positive in one patient, who had a titer of 240 KAU.
Table 3: Results of serological testing. View Table 3
A total of 37 patients (67.3%) received immunosuppressants. The individual agents received are shown in Figure 2. Of note patients may have received more than one immunosuppressant during their follow up.
 Figure 2: Treatment.
View Figure 2
Figure 2: Treatment.
View Figure 2
Symptom progress over the follow-up period from the time of diagnosis is shown in Table 4.
Table 4: Symptom progress. View Table 4
Follow-up CT scans were available in 48 (87%) patients, with the majority of patients showing no significant radiological changes between their first and last CT scans. UIP was the most common radiological diagnosis, being found in 24 (43.6%) patients. Other findings on HRCT included bronchiectasis changes, ground glass appearance and fibrosis. The HRCT findings for the cohort are shown in Table 5.
Table 5: CT findings. View Table 5
The pulmonary function test findings for the cohort are shown in Table 6. The minimum initial FVC was 0.70 L and the maximum was 3.74 L, while the corresponding results for the last FVC were 0.66l and 3.36l respectively. In terms of FEV1 the minimum first FEV1 was 0.69 L and the maximum was 3.09 L, while the minimum last FEV1 was 0.55 L, with a maximum of 2.93 L. We defined a difference of 10% between first and last pulmonary function tests as being required to categorise a patient's pulmonary function as having improved or deteriorated. However, if a difference of less than 10% were also considered, 34 (61.8%) patients had a deterioration in FVC, while 19 (34.5%) had an improvement in FVC. The median change in FVC was a decrease of 0.09l (p = 0.011). Similarly, the last FEV1 was significantly lower than the first FEV1 (median decrease 0.15 L, p = 0.001). Comparing individual pairs (not using the predefined 10% cut-off), the last FEV1 was lower than the first FEV1 in 39 (70.9%) cases, with an improvement in only 15 (27.3%).
Table 6: Lung function test trends. View Table 6
A total of 43 patients had 6-minute walk tests performed. The median initial 6-minute walk test was 430 m (IQR 350 m-487 m). Of these patients 31 had a subsequent 6-minute walk test. The median distance for the final 6-minute walk test was 420 m (IQR 360 m-470 m). The difference between first and last 6-minute walk test was not statistically significant (p = 0.189). Of note 20 (64.5%) patients had a deterioration in 6-minute walk test, while 11 (35.5%) had an improvement.
The diffusing capacity of the lungs for carbon monoxide (DLCO) was measured at least once in 34 patients, with 13 having a repeat test. The median initial DLCO was 3.5 L (IQR 3.1-4.5), last DLCO 3.4 L (IQR 3.1-3.9) and difference between first and last DLCO -2.1% (IQR -9.4%-4.1%). Of the patients with more than one DLCO results, 9 (69.2%) had no change, 3 (23.1%) had a deterioration in DLCO and 1 (7.7%) had an improvement in DLCO.
The median ESR at first presentation was 35 (IQR 20-60) while the median CRP was 7.0 (IQR 2.0-13.4).
The prevalence of ILD in our study was 37.6% which is similar to that described in another South African study from Gauteng Province (40%) and in studies from Europe (~35%) but may be lower than in studies from North America (~52%) [14,15]. These findings suggest that ILD is common in patients with SSc in Sub-Saharan Africa and all patients with SSc should be screened for pulmonary involvement.
The median age at time of diagnosis of ILD was 51 years in our cohort, which is slightly older than that reported in the other South African cohort (44 years) [14]. Our median age was however broadly similar to that seen in international studies. A US cohort study from 1997 to 2013, including 156 SSc-ILD patients, reported a mean age at diagnosis of 54.5 (± 13.2) years [16], which was consistent with results from two longitudinal US cohort studies assessing 225 SSc-ILD patients (mean age: 54 years) [17]. Similarly, the median age of SSc patients with severe ILD, identified as patients with an FVC < 60% from the pulmonary hypertension assessment and recognition of outcomes in scleroderma (PHAROS) registry in the United States and Canada, was estimated to be 52.5 years [18].
The majority (87%) of patients in this study were female, which is in keeping with global findings. Most of the patients (56%) in this study were, however, of Indian ethnicity, which is contrary to what has been described in other studies. The most likely explanation is the high Indian population in KwaZulu-Natal and Durban in particular. The Indian/Asian population makes up 24% of the population of Durban, and 8% of the population of KwaZulu-Natal [19]. This requires further study as genetic predisposition may have a role. In the study from Gauteng, the majority of patients were female (87.4%) and of black ethnicity (86.8%). Caucasians represented 2.7%, Indians 3.3% and mixed race individuals 7.2% [14]. Internationally similar gender ratios were observed in patients with SSc and SSc-ILD, with a female predominance in both Europe (75-82%) and North America (≥ 69%). Data from North America suggest that SSc-ILD and associated complications are observed most frequently in Caucasian patients [15].
In our study a significant proportion of patients had overlap with other connective tissues diseases. Systemic lupus erythematosus had the highest incidence of 18%, which may be due to the high prevalence of SLE in South Africa. There is limited data about the prevalence of other connective tissue diseases in patients with scleroderma ILD. In an Israeli study [16] examining the incidence of other connective tissue diseases in scleroderma patients (including forty patients who satisfied the criteria for scleroderma overlap syndrome), the incidence of additional connective tissue diseases in the whole group and in the overlap group respectively was 11.5% and 47.5% for dermatomyositis and polymyositis, 10.3% and 42.5% for Sjogren's syndrome, 3.6% and 15.4% for rheumatoid arthritis and 1.2% and 5% for systemic lupus erythematosus).
We found that most patients presented with skin thickening and Raynaud's phenomena, 49 (89.1%) and 46 (83.6%) respectively. This is similar to the Gauteng study which reported Raynaud's phenomenon (82.8%), and nailfold capillary changes (70.2%) as common presenting symptoms. In our study 47 patients (85.4%) presented with dyspnea, with most (58.2%) having Grade II dyspnea at the time of presentation. This may indicate that dyspnea is the best symptom marker of the development of ILD and can be used as a good and cheap screening tool.
ANA was positive in 50 of 54 patients (92.6%), anti-Scl-70 antibodies were positive in 21 of 45 (46.7%) patients and anti-centromere antibodies were positive in only 1 of 14 patients (7.1%). In the Gauteng study the ANA was positive in 88.1% of cases, with the commonest ANA patterns being speckled (40.6%), and nucleolar (31.6%). Only 7.5% had a positive ACA, while 19% were anti-topoisomerase positive [14]. This may indicate that the development of ILD in systemic sclerosis is more likely in patients with positive anti-Scl-70 and negative anticentromere antibodies. This is similar to what has been found in international studies where the variables that predicted clinically significant pulmonary fibrosis development were dcSSc, older age at onset, lower forced vital capacity and DLVO, and the presence of anti-Scleroderma-70 antibody. The presence of anticentromere antibodies was protective [17]. Anti-Scl-70 antibodies are also associated with worse prognosis, while ACA is associated with better prognosis [18].
In our study the majority of patients received cyclophosphamide as the main immunosuppressant for induction therapy followed by azathioprine. None in our cohort received any biological therapy, stem cell therapy or lung transplantation. This was due to resource limitations. In the Gauteng study corticosteroids were used in conjunction with cyclophosphamide as induction therapy followed by maintenance with either azathioprine or mycophenolate mofetil in 59.3% of ILD patients. Internationally in the EUSTAR study (which included 3778/11,496 patients with SSc-ILD (33%)), glucocorticoid (GC) monotherapy was prescribed in 30.6% of patients, GC combinations plus cyclophosphamide (CYC) in 11.9%, azathioprine in 9.2%, methotrexate in 8.7%, and mycophenolate mofetil in 7.3% [6]. The regimen used in our study compares favorably with regimens used elsewhere. Of note, in our cohort corticosteroids were not included. MMF was less frequently used as induction therapy despite a more favorable side effect profile compared to cyclophosphamide. The reasons for this need to be interrogated further, with consideration given to future use of MMF.
The most common finding on HRCT in our study was a UIP pattern. This may be due to late presentation of patients. This is dissimilar to the international literature where typical CT findings were ground glass opacification with an admixture of pulmonary fibrosis consistent with the NSIP pattern. Honeycomb cystic changes are reported in 11% to 37% of patients with scleroderma-ILD [20]. This is unlike other patients with NSIP, who have little or no cystic change. As honeycomb cystic change is typically a marker for UIP and pulmonary fibrosis. These findings suggest that patients with scleroderma-ILD disease may have a mixture (or overlap) of UIP and NSIP patterns.
When assessing longitudinal impact on pulmonary function using a change in FVC of 10%, most patients remained static, with approximately one third deteriorating. In a study that tracked lung function over a mean of 6.4 ± 4.2 years, patients underwent a median of 5 PFTs during the follow up [21]. While there was no significant decline in FVC overall during the study period (-0.1 ± 0.3%/year, p = 0.71), 29% of patients experienced a significant decline of FVC of more than 10% from baseline after 6 years of follow up. In another study of 105 patients (45 diffuse SSc; median disease duration 2.0 years), 23 (23%) had an FVC of < 80% predicted, and 60 (59%) had a DLCO of < 80% predicted. Over 72 ± 46 months, 29 (28%) patients displayed a decrease of ≥ 10% in FVC, 39 (40%) of 98 patients displayed a DLCO decline and 19 (18%) patients displayed a decrease of ≥ 20% in FVC. On multivariate analysis, diffuse SSc was a significant predictor for a decrease of ≥ 10% in FVC (p = 0.01) [22]. This shows that the majority of patients remain static or improve. Maintaining lung function is a positive outcome.
This was a retrospective review of electronic data and only data captured and available could be analysed. It was a small sample size. The small sample size as well as missing data may have biased any associations. However, scleroderma-ILD is a disease with a very low prevalence; hence smaller sample sizes are to be expected. There is the possibility of overestimation of prevalence as the study was conducted in a specialized clinic in the major referral hospital for the province. This may have resulted in referral bias. There may also have been patients with interstitial lung disease, who were missed because they were asymptomatic. Testing and follow-up intervals were not standardised which is another potential source of bias. Patients had different durations of follow up. Repeat HRCT scans were based on clinical judgement by the treating physician and was not standardized.
This is the second study, to our knowledge, in South Africa, to report on the demographics and treatment of scleroderma-ILD in a multiracial cohort. While age and gender distribution are consistent with global data, there is apparently a high prevalence of scleroderma-ILD among South Africans. UIP is the dominant radiological pattern. Cough and dyspnea could be used as good screening tools for early detection of scleroderma-ILD. The use of cyclophosphamide and azathioprine appears to prevent disease progression. Increased utilization of MMF should be considered. Further prospective multi-centre randomized controlled trials are required in this field to determine the optimal drug treatment to manage Scleroderma-ILD.