Adult stem cells hold tremendous potential for regenerative medicine. Harnessing their therapeutic benefits requires that we gain fundamental understanding of the mechanisms by which stem cells function and divide in vivo. This is hampered by the fact that stem cells often reside in compartments that remain difficult to access using imaging approaches. The nematode C. elegans is transparent and studying its germ line stem cells (GSCs) offers a powerful means to characterize how stem cells divide in vivo. In this review, we present an overview of the core molecular events that govern GSC division in C. elegans and describe what lessons can be learned from these cells to further understand adult stem cell division in other systems.
Adult stem cells, Germline stem cells, Caenorhabditis elegans, in vivo stem cell regulation, Niche signaling
Pioneering work by Till and McCulloch, in the early 1960s demonstrated the existence of multipotent stem cells in the mouse bone marrow that could self-renew and differentiate into other cell types [1,2]. This capacity to both self-renew and differentiate is a hallmark that characterizes all known stem cells (SCs). Classic blastomere transplantation experiments performed in the early mouse embryo in the 1970s defined two additional kinds of stem cells: totipotent SCs and pluripotent embryonic SCs [3]. Totipotent SCs produce all the differentiated cells in an organism, including extra embryonic tissues, while embryonic SCs (only present in mammals) can differentiate into all three germ layers and produce the embryo proper [4]. Multipotent SCs, also known as tissue-resident or adult SCs, on the other hand, have a more restricted differentiation potential, and can only give rise to one or more related cell types. Whereas totipotent SCs and embryonic SCs only exist in the early embryo, adult SCs persist over the entire life of an organism, thereby ensuring the normal turnover and/or repair of adult tissues. Depending on various factors, including tissue demand, adult SCs can proliferate, differentiate, or remain quiescent. Since adult SCs collectively possess the ability to regenerate most adult tissues, they offer major therapeutic potential.
The introduction of in vitro SC culture revolutionized SC research by facilitating the expansion, manipulation (genetic and pharmacological) and microscopic observation of SCs [5]. Yet, SCs in vivo respond to various signals provided by a specialized somatic cellular environment termed the niche, which is not fully recapitulated by in vitro culture conditions. SCs are also sensitive to growth factors whose concentrations can vary during development or can be affected by environmental conditions. Our current understanding of the role of the SC niche, which signals to SCs to maintain their undifferentiated state, stems from pioneering studies in invertebrate models, such as Drosophila [6,7] and C. elegans [8]. SC niches were later identified in vertebrates and are conceptually very similar to invertebrate niches, including how they signal to the SCs [9-11]. Furthermore, the study of SC sensitivity to environmental and physiological factors was initiated by a key contribution from Drummond-Barbosa and Spradling, who demonstrated that the proliferation of Drosophila germline SCs (GSCs) responds to nutritional input [12]. Their work revealed that diet controls GSC proliferation by influencing the secretion of insulin-like peptides by neuro-secretory cells in the Drosophila brain and thus changing insulin/IGF-1 signaling levels [12,13].
The study of SCs in vivo has been hindered by their limited accessibility in most vertebrate systems, which require surgical dissections and/or complex transplantation assays [3]. Recent advancements in intravital imaging of certain murine adult SC populations have yielded invaluable insights into how SCs interact with their niche and alter their behaviour in response to injury [14]. This work highlights the importance of studying SCs within their native environment and the need for additional model systems that effectively combine genetic, cell biological and molecular approaches to the study of SCs in vivo.
The nematode C. elegans has been used for the last 50 years to uncover and characterize gene function within an in vivo context. C. elegans animals are transparent, which permits visualization of all cellular processes in situ, without the need for surgical manipulation (Figure 1A). The gonad of adult C. elegans hermaphrodites forms two symmetrical U-shaped arms in which ∼1000 syncytial germ cells are organized in a columnar monolayer around a common central cytoplasmic core, known as the rachis [15]. Each gonad arm is polarized, from distal to proximal, with germ cells progressing through all stages of meiosis, culminating with fully differentiated gametes at the proximal end, near the spermatheca, where oocytes are fertilized and extruded into the uterus (Figure 1B). The distal-most region of each gonad arm contains a population of GSCs that undergo self-renewing mitotic divisions (Figure 1C), but can also differentiate to give rise to terminally differentiated gametes (sperm and oocytes). In this sense, C. elegans GSCs essentially recapitulate the features of adult SCs found in mammals.
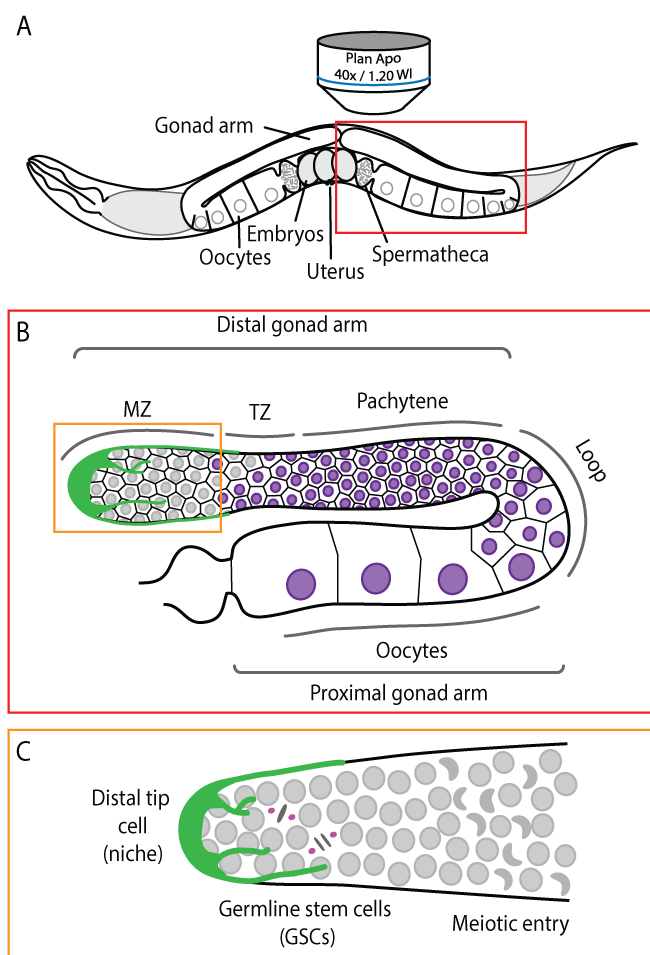 Figure 1: The C. elegans germline. (a) Schematic depiction of an adult hermaphrodite worm. The germline is readily accessible for live cell microscopy in whole mount animals; (b) Enlarged schematic depiction of a single gonad arm. Germ cells are arranged in a conveyor belt-like fashion with mitotic germ cells at the distal end of the gonad arm (the mitotic zone, MZ). As cells move along the distal arm they enter meiosis in the transition zone (TZ) and progress through pachytene. In the loop region, the majority of germ cells undergo apoptosis and the remaining cells begin to enlarge and mature into oocytes, which occupy the proximal arm; (c) Enlarged schematic depiction of the mitotic zone. GSCs divide mitotically to maintain the size of the GSC pool while also sustaining oocyte production. GSCs enter meiosis as they leave the niche, forming crescent-shaped nuclei. The niche (the distal tip cell) surrounds the distal end of the gonad arm and extends processes proximally along the mitotic zone. For simplicity, plasma membranes were omitted in panel.
View Figure 1
Figure 1: The C. elegans germline. (a) Schematic depiction of an adult hermaphrodite worm. The germline is readily accessible for live cell microscopy in whole mount animals; (b) Enlarged schematic depiction of a single gonad arm. Germ cells are arranged in a conveyor belt-like fashion with mitotic germ cells at the distal end of the gonad arm (the mitotic zone, MZ). As cells move along the distal arm they enter meiosis in the transition zone (TZ) and progress through pachytene. In the loop region, the majority of germ cells undergo apoptosis and the remaining cells begin to enlarge and mature into oocytes, which occupy the proximal arm; (c) Enlarged schematic depiction of the mitotic zone. GSCs divide mitotically to maintain the size of the GSC pool while also sustaining oocyte production. GSCs enter meiosis as they leave the niche, forming crescent-shaped nuclei. The niche (the distal tip cell) surrounds the distal end of the gonad arm and extends processes proximally along the mitotic zone. For simplicity, plasma membranes were omitted in panel.
View Figure 1
The complete lineage analysis of C. elegans was done more than 30 years ago, allowing a detailed understanding of cell differentiation and interactions during development. All germ cells originate from a single germline blastomere, termed P4, that is born during embryogenesis out of a succession of asymmetric cell divisions [16,17]. Around the 100-cell stage, the P4 blastomere divides symmetrically into two primordial germ cells that remain mitotically quiescent during the remainder of embryogenesis. The primordial germ cells resume mitotic proliferation during the first larval stage and the germline undergoes proliferative expansion throughout the following three larval stages [15]. Proximal germ cells enter meiotic differentiation during the third larval stage to generate gametes during the fourth larval stage and adulthood, first forming ∼300 sperm then switching to oocyte fate. Adult hermaphrodites maintain two pools of ∼200 GSCs that undergo homeostatic self-renewal, one at each distal extremity of the two gonad arms (Figure 1).
Laser ablation experiments performed in the early 1980s revealed that a single somatic cell, known as the Distal Tip Cell (DTC), acts as niche for C. elegans GSCs [8]. In hermaphrodites, two DTCs are born during the first larval stage and, in addition to promoting GSC fate, migrate throughout larval development to elongate and structure each gonad arm. When migration stops, the two crescent-shaped DTCs start elongating membrane processes toward the proximal end of the gonad [18,19], although the precise role of DTC elongation remains unclear.
Thorough molecular and genetic analyses revealed that the DTC relies on a Notch ligand, termed LAG-2, to activate the Notch receptor at the surface of GSCs [20,21]. Activation of Notch signaling in GSCs results in the induction of mitotic fate regulators and the repression of meiotic fate regulators, thus promoting GSC self-renewal over differentiation [22]. This is an active process as ablating the DTC or mutating the Notch receptor both result in loss of GSC fate and entry into meiosis, while displacing the DTC or over activating the Notch receptor causes GSC over proliferation, a phenotype reminiscent of benign tumors in mammals [8,20,23,24]. In wild-type animals, germ cell meiotic entry therefore naturally occurs when GSCs move away from the DTC and hence exit the niche, presumably as a consequence of sustained GSC proliferation.
Recent evidence suggests that all pre-meiotic germ cells are equivalent and only vary in their degree of intracellular Notch activity, depending on the amount of signal they receive from the niche, which is inversely proportional to their distance from the DTC body [25]. Importantly, G2 GSCs that stop receiving niche signaling undergo one single mitotic division before entering meiosis and as such there is no transit amplification prior to differentiation in this system [26]. Undifferentiated germ cells, which may encompass both stem and progenitor cells, are thus collectively referred to as GSCs.
Similarly to mammalian adult SCs, C. elegans GSCs exhibit quiescence, during which they temporarily arrest cell cycle progression while remaining in an undifferentiated state. The decision to proliferate or enter quiescence is made through integration of multiple signals. Following embryogenesis, after the animals have begun to feed, a neuronal signal activates insulin/IGF-1 signaling (IIS) to initiate larval growth and the two formerly quiescent primordial germ cells begin mitotic proliferation [27]. The GSC pool undergoes proliferative expansion during larval development, peaking at about 200 GSCs per gonad arm in early adulthood, and then slowly decreasing in number as animals age [28-31]. Expansion of the GSC pool and maintenance of its size is dependent on nutrient uptake which, through IIS levels, stimulates GSC proliferation in larvae and adults [29,32,33]. Down regulation of DAF-2, the sole C. elegans insulin/IGF-1 receptor ortholog [34], triggers the activation and nuclear translocation of the forkhead transcription factor DAF-16/FOXO [35,36], which inhibits GSC proliferation by activating the transcription of several stress response genes in the germ line [23,33,37]. Poor nutrition and/or reduced IIS therefore leads to adult animals having a lower number of GSCs per gonad arm [31,38]. In addition to being influenced by IIS levels, the size of the GSC pool appears to be controlled by sensory inputs through TGF-β signaling levels in the niche [39].
The interpretation of changes in GSC pool size can be confounding, as the adult GSC pool size depends on a combination of proliferation during larval growth, extent of niche signaling and the propensity of GSCs to differentiate. However, information on past GSC proliferation can be inferred through measurements of the GSC pool size during the early stages of development, before the proximal-most GSCs have begun to differentiate [28,32,40]. Such measurements revealed that a decrease in IIS results in reduced GSC proliferation during larval development [32-33], thus potentially contributing to the smaller GSC pool size found in daf-2 mutant adults.
The lack of nutrition and/or the resulting down regulation in IIS is not the only factor that can promote GSC quiescence. When they encounter harsh environmental conditions and/or over-crowding early in their development, C. elegans larvae can enter a diapause-like stage called dauer during which GSCs enter G2 quiescence [32]. While starvation promotes dauer formation via IIS down regulation, overcrowding inhibits TGF-β signaling and the inactivation of either pathway is sufficient to induce dauer formation and GSC quiescence [32,41]. When dauer formation is induced through a down regulation of TGF-β signaling, DAF-16/FOXO is dispensable for the installment of GSC quiescence [32]. Similarly, larvae hatched in the absence of food do not require DAF-16/FOXO to maintain GSC quiescence [40]. The key gene products that couple nutritional inputs and sensory information to GSC activity independently of DAF-16/FOXO in larvae include C. elegans orthologs of the dual specificity phosphatase PTEN, and the protein kinases LKB1 (along with its co-factor STRADα) and AMPK [32,40,42,43]. While the molecular link between PTEN and LKB1 remains obscure, it is clear that LKB1 activates AMPK through direct phosphorylation on a conserved residue in AMPK's activation loop [42,44]. AMPK is also allosterically stimulated by AMP, the concentration of which rises under energetic stress [45]. These proteins are very well conserved throughout eukaryotes and their homologs have since been reported to similarly influence SC proliferation in mammals. Indeed, PTEN [46,47] and more recently, LKB1 [48-50] were both found to be necessary for the maintenance of murine hematopoietic SC quiescence. While very recent work also confirms a contribution of AMPK in the regulation of hematopoietic SC activity [48,51], its precise role remains unclear. Fragmented evidence, in both C. elegans and mammalian systems, suggests that PTEN, LKB1 and AMPK inhibit SC growth and proliferation largely through inactivating the mTOR complex in SCs, thus decreasing protein synthesis [48,52-55].
Proliferation rates directly report on GSC stimulation at any given time and can be evaluated in vivo by various means, including mitotic index (MI) or S-phase index measurements [30,56]. Using such methods, it was found that GSC proliferation in adult C. elegans is further influenced by nutrition-independent factors. In hermaphrodites that are sperm depleted, oocytes arrest their maturation and accumulate in the proximal gonad [57]. Oocyte accumulation promotes G2 GSC quiescence, which is perturbed by rare, stochastic and transient bouts of proliferation, leading to an overall drastically reduced proliferation rate [29,58]. As is the case under certain circumstances during larval development, the promotion of GSC quiescence in adult hermaphrodites that are devoid of sperm is dependent on PTEN, but independent of DAF-16/FOXO [29]. Most interestingly, the regulation of GSC proliferation through this process occurs independently in the two gonad arms, thus effectively coupling each GSC population to the needs of its own gonad arm for oocyte production [29]. This is accomplished despite a systemically activated IIS pathway that would otherwise promote GSC proliferation equally in both gonad arms. These results indicate that PTEN acts to locally antagonize systemic IIS and block GSC proliferation in order to couple GSC proliferation to oocyte needs [29].
Such localized adult SC regulation is unlikely to be restricted to C. elegans GSCs. For example, mammalian muscle SCs, that are normally quiescent throughout adult life, resume proliferation to sustain muscle repair following injury [59,60], and they achieve this without any nutritional change. Furthermore, as no systemic muscle overgrowth has been reported following localized injury, it is likely that only the muscle SCs located close to the injury resume proliferation. The C. elegans GSCs therefore constitute an excellent model for the study of adult SC activity regulation, and we anticipate that much more will be learned in this system regarding the regulation of adult SC activities, including by systemic information, by aging, and by tissue demand.
All SCs, irrespective of tissue type, share one common attribute-they must, at some point, divide mitotically. Mitosis drives developmental expansion of the stem cell pool, sustains homeostatic self-renewal and permits tissue regeneration upon injury. Several features of SCs may impose specific requirements on how they undergo mitosis. The dependence of SC identity on proximity to the niche means that the orientation of cell division, which will impact daughter cell placement relative to the niche once division is complete, may be important for maintaining a balance between SCs and their differentiating progeny. Spindle orientation in SCs may be regulated such that each SC division is asymmetric, i.e., one daughter cell remains adjacent to the niche, while the other is displaced, as is the case for GSCs in the Drosophila testis [61]. Regulated spindle orientation can also be subject to orientation switching. Drosophila female GSCs can divide both asymmetrically and symmetrically relative to the niche, with the former producing differentiating cystoblasts and the latter replacing a neighbouring stem cell [62]. Alternatively, in tissues where SCs are maintained at the population level, cell division is symmetric with respect to cell fate (i.e., both daughter cells have the potential to be SCs or to differentiate) and spindle orientation relative to the niche may be randomly distributed. A fixed SC pool size is then maintained by stochastic loss of SCs from the niche, a mode of SC maintenance termed population asymmetry that appears common in rapidly cycling tissues such as the intestine and epidermis [63].
C. elegans GSCs are likely to follow a model of population asymmetry [11]. During germline development, GSC divisions are thought to be symmetric, as the orientation of division appears variable, ablation of one or more GSCs is compensated for by surviving cells and ectopic positioning of the niche is sufficient to induce GSC fate [8,64]. In adult GSCs, spindle orientation relative to the niche also appears widely variable with divisions both parallel and perpendicular to the proximal-distal (niche) axis [30]. However, we note that adult niche morphology is remarkably complex, with long, branching processes extending many cell diameters away from the DTC body [18,19]. The recent development of live imaging strategies for the direct observation of GSC divisions [65] will facilitate future inquiries into the function of these niche extensions and whether they impact spindle orientation in GSCs.
SCs contribute to tissue homeostasis over remarkable time scales, from days to years. Thus maintaining genome fidelity upon SC division is critical to their function. A key regulator of mitosis that is responsible for preserving mitotic fidelity is the spindle assembly checkpoint (SAC). During mitosis, replicated chromosome pairs connect to opposite spindle poles and become aligned on the metaphase plate. This bi-orientation guarantees that upon anaphase chromatid separation, each daughter cell receives the correct chromosome number [66]. Attachment of sister chromatids to the mitotic spindle is an inherently stochastic process of variable duration; thus, to prevent chromosome segregation errors and maintain genome stability, anaphase onset is blocked by the SAC until bi-orientation is achieved [67]. Weakened SAC function leads to aneuploidy and has been associated with tumor development in both model systems and humans [68]. Addressing SAC activity in SCs has been difficult, as thorough assessment of mitotic progression and accurate timing of events such as anaphase onset require live imaging approaches, which are challenging for most SC populations. As C. elegans GSCs are amenable to live imaging studies, they have provided an opportunity to investigate how the SAC functions in SCs. Notably, compared to embryonic blastomeres, which have a relatively weak SAC [69-71], GSCs show enhanced SAC activity with longer SAC-dependent mitotic delays following spindle perturbations [65]. Determining at a molecular level how these changes are achieved over the course of germline development and how they may be linked to SC fate is an interesting area for future inquiry.
Lastly, another feature of SC mitosis derives from the location of these cells in mature tissues within the broader physiological environment of an adult organism. To sustain tissue function over the lifetime of an organism, SCs must execute mitosis under a range of physiological conditions. While various signals can induce a G1 or G2 cell cycle arrest, including unfavourable environmental conditions, within the complexity of a living organism, SCs are likely to encounter a range of suboptimal conditions, which are not necessarily inhibitory for cell cycle progression. Moreover, in tissues with a high rate of cellular turnover, such as the intestine or epidermis, complete cell cycle arrest may interfere with tissue function, as is evidenced by the negative side effects of many anti-proliferative cancer drugs. Thus SC mitosis is likely to be subject to a range of physiological and environmental inputs. Live imaging of C. elegans GSCs has revealed that key mitotic parameters, including the timing of anaphase onset, are affected by physiological manipulations, such as dietary restriction, and developmental changes, such as the transition from developmental expansion to adult maintenance of the SC pool [65]. Dietary restriction, in particular, appears to impair chromosome congression and/or spindle assembly, as eliminating the SAC in calorie-restricted animals caused an increase in chromosome segregation errors relative to well-fed controls [65]. The mechanism by which dietary restriction impacts these core mitotic processes remains unknown and, importantly, whether SAC activity is up regulated to compensate for impaired spindle assembly when diet is restricted has not been established.
C. elegans GSCs thus constitute an outstanding and uniquely accessible model system to study in vivo SC regulation at all levels, including how they are regulated by the niche, how their activity is influenced by growth factors, and how mitotic accuracy is ensured. The importance of understanding how adult SC division is regulated in vivo is bolstered by its relevance for regenerative therapies. For example, it may be beneficial to stimulate a given population of skin and/or muscle SCs to enhance healing in an elderly patient, or of neural SCs to overcome a neurodegenerative disorder. However, understanding which molecules are specifically involved in locally coupling the activity of the targeted adult SC population to its differentiated progeny needs will require further investigation in order to envisage such a strategy. In light of recent results in C. elegans and Drosophila, it would indeed appear unwise to try to accomplish adult SC reactivation by systemically stimulating SC activity, for example through activating IIS. The effects of this would be systemic and hence have undesired side-effects in untargeted tissues, but more importantly, they could be abolished by local feedback signals originating from the differentiated SC progeny [29]. Further genetic and molecular characterization of this mechanism using C. elegans GSCs may help to guide experimental exploration of this process in mammalian systems.
A better understanding of in vivo SC regulation may also be desirable for cancer prevention and treatment, as evidence suggests that cancer may arise from defects in stem cell division. Indeed, a recent study has uncovered a strong positive correlation between the number of times SCs divide over a person's life in a given organ and the lifetime cancer risk for that organ [72]. While age has long been considered as the greatest risk factor for developing cancer [73], this correlation suggests that the greatest cancer risk factor may actually be the total number of divisions stem cells execute over our lifetime. This view is strengthened by the current mechanistic understanding of tumorigenesis, which is believed to arise following accumulation of tens to hundreds of inherited genetic and epigenetic changes that occur in cycling cells and collectively favour cell proliferation and survival [74-76]. Thus, it would seem reasonable to hypothesize that the older we live, the more our SCs divide, the more they can progress towards unregulated growth and eventually, tumorigenesis. Understanding what normally restrains adult SC proliferation in vivo, and how the mechanisms that ensure the accuracy of SC divisions are implemented, could therefore help us to better define the initial steps that underlie tumorigenesis.
Finally, we note that the C. elegans GSCs follow a model of population asymmetry, which is also associated with tissues that undergo rapid homeostatic turnover in human adults, including the intestinal epithelium. Interestingly, germline mutations in the human orthologs of some of the genes required for GSC quiescence in C. elegans (PTEN and LKB1) lead to the related dominantly-inherited tumour-predisposing Cowden's and Peutz-Jegher's syndromes, respectively [77-79]. Incidentally, the main clinical features of these syndromes is the appearance of multiple benign polyps in the gastrointestinal tract [80]. Based on the similarity of this clinical feature to the C. elegans phenotype shown by mutants in these genes, and of their underlying cellular and molecular cause, we propose that the C. elegans GSCs may represent a particularly well-suited system to better understand these syndromes.
We thank members of the Maddox and Labbé laboratories for helpful discussions. This work was funded by a grant from the Canadian Institutes of Health Research (MOP-115171) to J-C.L. and P.S.M. IRIC is supported in part by the Canada Foundation for Innovation and the Fonds de Recherche du Québec -Santé.