Latent infection is a major barrier for cure of HIV-1 infection. HIV-1 is capable of establishing latency when its host cells undergo apoptosis and initiate replication in response to this effect. HIV infection results in increased p53 expression that enhances replication in primary infection. To examine whether p53 reactivates HIV-1 replication from latent infection, we used susceptible U1 cells chronically infected with HIV-1 to study the effects of p53 on reactivation of HIV-1 replication in the monocyte/macrophage cell reservoir. We found that p53 could reactivate HIV-1 replication from latency in U1 cells with upregulation/activation of host transcription factors, NF-κB, NFAT, AP1, SP1 and c-Fos, and through modulation of PI3K/Akt and MAPK Erk/p38 signaling pathways. p53 induced P-TEFb signaling with upregulation of CDK6 and Cyclin T1, and increased phosphorylation of the carboxyl-terminal domain of the large subunit of RNA polymerase II. We also found that p53 upregulated CD4, LEDGE/p74 and PARP-1 and downregulated COMMD1 in U1 cells. p53 expression resulted in the increased expression of acetyl transferase PCAF and decreased expression of de-acetylation HDAC1/2 and SIRT1, which facilitate access of RNA polymerase II to active chromatin to enhance HIV-1 replication. These findings indicate that p53 plays a positive role in reactivation of HIV-1 replication from its latent state at different levels including upregulation/activation of host transcription factors and recruitment of Histone Acetyl Transferase (HAT)/inhibition of histone deacetylation to acetylate histone tails and to open nucleosomes to facilitate HIV transcription.
Macrophage, HIV, p53, Transcription factor, Epigenetic factor
Human Immunodeficiency Virus type-1 (HIV-1) infection of humans causes progressive destruction of CD4+ T cells, as well as chronic immune activation resulting in immune exhaustion and depletion of uninfected immune cells. The use of effective combined Anti-Retroviral Therapy (cART) has improved duration and quality of life of people living with HIV-1 [1]. However, the virus continues to persist in long-lived resting CD4+ T cells, macrophages and astrocytes which form a viral reservoir in infected individuals that impede complete eradication of the virus [1]. Currently, all treatment efforts aimed at curing HIV-1 infection have been mainly targeted against latently infected resting memory CD4+ lymphocytes [2]. To cure HIV-1 infection, it is essential to consider all possible long-term HIV-1 reservoirs harboring either the latent or active form of the virus. Technical and biological issues have hampered a better understanding of non-T cell reservoirs [3]. A recent study using a humanized mouse model shows that HIV-1 can persist in macrophages during antiretroviral therapy, and suggests that macrophages may represent an obstacle to efforts to cure HIV-1 infection [4].
p53 is known as a tumor suppressor factor and its gene is located on the short arm of chromosome 17 (17p13.1). As a regulator of cell proliferation, apoptosis and senescence, p53 has expanded greatly to be involved in many biological processes as well as external and internal stress responses [5]. p53 plays important roles in immune-related processes such as inflammation, innate and adaptive responses as well as functional interactions of p53 with NF-κB, which is considered a key regulator in immune responses [5]. HIV-1 infection is able to induce cytotoxic stresses linked to p53 activation in CD4+ T cells by integration mediated-dsDNA strands break [6].
Transcription of HIV-1 provirus in infected cells is regulated by the interaction of both viral proteins and cellular transcription factors with the viral Long Terminal Repeat (LTR) sequences [2]. The HIV-1 LTR has been shown to contain recognition sequences for many cellular transcription factors, including NFAT, NF-κB, AP-1 and SP-1 [7-9]. Accumulating evidence suggests that early HIV-1 gene products, such as Nef and Tat, promote NF-κB activation to initiate productive viral replication. In the presence of Tat, the HIV-1 LTR is highly active even when only low levels of activated NF-κB and other transcription factors (such as NFAT, AP-1, SP-1) are present [7-9].
One approach, termed as "shock (or kick) and kill," is to deplete the reservoir by activating latent HIV while using cART to prevent infection of new cells [10]. The agents used in this approach that have been studied, so far, include those involved in activation of NF-κB pathway and PKC signaling, alteration of epigenetic environments (such as DNA methylation inhibitors, and histone deacetylase inhibitors) [10]. Latent viruses also can be activated at the level of elongation of transcription. The recruitment of P-TEFb (positive Transcription Elongation Factor b by Brd4 (Bromodomain containing protein 4) during the elongation stage is also important for the full length synthesis of HIV-1 mRNA, which is manipulated by the Tat protein essential for viral replication. The protein cyclin T1, tightly associated with CDK9 (Cyclin-Dependent Kinase 9) to form a major subunit of P-TEFb [11]. The CDK9/cyclin T1 complex is an important factor for productive elongation of transcription of HIV genomes, and the inhibition of CDK9 can ameliorate HIV‐induced disease in animal models [11]. In cultured human T cells, dominant negative CDK9 mutants impair HIV‐1 replication. HMBA (Hydroxy Methyl Benzoic Acid), a prototype hybrid polar compound, is able to activate P-TEFb and its recruitment factor, bromodomain containing protein Brd4, leading to the activation of transcriptional elongation of HIV-1 [11].
Cumulative evidence has shown that virus enhances its replication when host cells initiate the apoptotic program. This phenomenon is called an Alternative Replication Program (ARP), a process when the host cell is undergoing apoptosis during viral infection. Viruses adopt an emergency escape mechanism to produce a large amount of virus rapidly [12]. Expression/activation of some pre-apoptotic molecules from apoptotic pathways, such as p53, Bax, Fas ligand, FADD, caspase-3, and caspase-8 are sufficient to initiate the ARP in HIV-1 infection [13] and influenza A virus infection [14]. Treatment with cytotoxic drugs has been reported to produce and activate HIV-1 replication in pro-monocytic (U1) and lymphoid (ACH-2) cell lines persistently infected with HIV-1 with an increased expression of p53 [12]. Here using U1 cells, we found that p53 expression was able to activate HIV-1 replication from latent infection via upregulation of several host transcription factors in U1 cells.
Rabbit polyclonal antibodies against AKT, Brd4, CD4, CDK9, c-Fos, p53, PARP-1, PI3K, SP1 and GAPDH were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). AP-1, COMMD-1, Cyclin T1, ERK, EZH2, HDAC1, HDAC2, HEXIM1, JNK, LARP7, LEDGF/p75, p24, p38, PCAF, p-Rpb-CTD, NFAT, NF-κB p65, and SIRT1 were bought from Cell Signaling Technology, Inc (Danvers, MA). The full-length gene p53 or LacZ inserted in an adenovirus expression system were obtained from the Molecular Medicine Institute Programs of Excellence in Gene Therapy Vector Core Facility, University of Pittsburgh. HMBA and all other chemicals were from Sigma (St. Louis, MO).
U1 (promonocytic) cells, a subclone of U937 chronically infected with HIV-1, were obtained from the National Institutes of Health AIDS Research Reference and Reagent Program (Germantown, MD) and cultured at 37 ℃ in 5% CO2 in RPMI 1640 medium containing 10% fetal calf serum, 2 mM glutamine, 50 μg/ml penicillin, and 50 μg/ml streptomycin. For adenoviral infections, U1 cells were seeded at 2 × 105 cells/ml for 24 h, and infected with 108 CPU/ml of an adenoviral vector inserted with a gene for 2 h, washed twice with PBS and cultured for 3 days with presence/absence of 5 mM of HMBA.
Quantitative real-time Reverse-Transcriptase (RT) PCR was used for quantitation of viral RNA. Viral RNA was isolated from 140 µl of culture supernatant by using the QIAamp Viral RNA Mini Kit (Valencia, CA 91355) according to the manufacturer's protocol. The primers and TaqMan probe were designed in the gag capsid (p24) region, which is the variable region among most of the HIV-1 subtype B isolate sequences according to GenBank database. The forward primer was 5'-CTATCCCATTCTGCAGCTTCCT-3', corresponding to nucleotides 1367-1386, and the reverse primer was 5'-ATTGATGGTCTCTTTTAACA-3', corresponding to nucleotides 1430-1409. The TaqMan probe was oligonucleotide 5'-ATTGATGGT CTCTTTTAACA-3', corresponding to nucleotides 1488-1507, coupled with a reporter dye [6-carboxyfluorescein] (FAM) at the 5' end and a non-fluorescent quencher and a Minor Groove Binder (MGB), which is a Tm enhancer, at the 3' end. The nucleic acids were amplified and detected in an automated TaqMan 7500 Analyzer by using QuantiTect™ Probe RT-PCR kit (Qiagen Inc., Valencia, CA). The 25-µl PCR mixture consisted of 100 nM primers and 100 nM probe. Following three thermal steps at 55 ℃ for 5 min, at 50 ℃ for 30 min and at 95 ℃ for 10 min, 45 cycles of two-step PCR at 95 ℃ for 15 s and at 60 ℃ for 1 min were performed.
The data are expressed as copy numbers/ml. Known concentrations of HIV-1 (MN) viral RNA (serially diluted: 108 to 100 copies) were used as templates and quantitative RT-PCR performed to generate a standard curve. Each value represents the average concentration of six reactions in triple isolated repeats based on the standard curve.
Proteins were isolated from U1 cells with RIPA buffer (1 × PBS, 1% (v/v) NP-40, 0.5% (w/v) sodium deoxycholate, 0.1% (w/v) SDS, 0.1 mg/ml PMSF, 30 μl/ml aprotinin, 1 mM sodium orthovanadate). Equal amounts of protein were boiled in the loading buffer (100 mM Tris-HCl, 200 mM DTT, 4% SDS, 0.2% bromphenol blue, 20% glycerol), separated on SDS-PAGE and blotted onto polyvinylidene difluoride membranes. The data represented are from three independent experiments. The relative quantitation of protein expressions was determined with Image J (Image Processing and Analysis in Java) from the NIH website (https://imagej.nih.gov/ij/).
The unpaired Student's t test was used for data analyses as indicated, and a value of p < 0.05 (*) was considered significant, p < 0.01 (**) very significant.
We have reported that HIV-1 infection induces more p53 expression in Jurkat cells relative to HIV-2 infection [15] and p53 expression is able to increase HIV-1 replication in primary infection [13]. To study the effects of p53 expression on activation of HIV-1 latent infection in macrophages, U1 cells were infected with adenovirus containing an inserted gene for p53 or LacZ for 3 days and culture supernatants were used for RT-PCR and proteins from cell pellets were subjected to Western blotting analysis. As shown in Figure 1A and Figure 1B, U1 cells that expressed p53 (Figure 1A) displayed an increased HIV-1 capsid p24 expression (Figure 1A) and produced more HIV-1 virus relative to the LacZ control (Figure 1B), suggesting that p53 expression is able to reactivate HIV-1 replication from its latent infection.
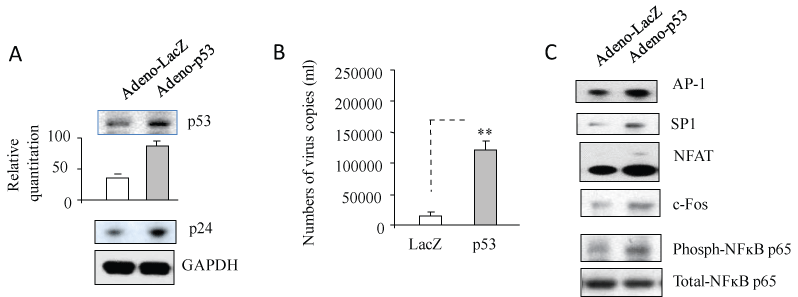 Figure 1: p53 expression increased the activation of HIV-1 replication through upregulation of host transcription factors. U1 cells were infected with adenovirus inserted a gene, p53 or LacZ for 2 hours, washed with PBS and then cultured for 3 days. After incubation for 3 days, (B) 140 µl of culture supernatants containing HIV-1 particles were used to isolate viral RNA. 10 µl in 50 µl of the RNA was used as template to perform real-time PCR. Known concentrations of HIV-1 (MN) viral RNA (serially diluted: 108 to 100 copies) were used as templates and quantitative RT-PCR performed to generate a standard curve. Each value represents the average concentration of six reactions in triple isolated repeats based on the standard curve. The value of p < 0.05 (*) was considered significant, p < 0.01 (**) very significant. The cell pellets were lysed with RIPA buffer and the cell lysates were subjected to Western blot analysis to detect to p53 and HIV-1 p24 (A) and NFAT, SP-1, AP-1, c-Fos and NF-ĸB (C). View Figure 1
Figure 1: p53 expression increased the activation of HIV-1 replication through upregulation of host transcription factors. U1 cells were infected with adenovirus inserted a gene, p53 or LacZ for 2 hours, washed with PBS and then cultured for 3 days. After incubation for 3 days, (B) 140 µl of culture supernatants containing HIV-1 particles were used to isolate viral RNA. 10 µl in 50 µl of the RNA was used as template to perform real-time PCR. Known concentrations of HIV-1 (MN) viral RNA (serially diluted: 108 to 100 copies) were used as templates and quantitative RT-PCR performed to generate a standard curve. Each value represents the average concentration of six reactions in triple isolated repeats based on the standard curve. The value of p < 0.05 (*) was considered significant, p < 0.01 (**) very significant. The cell pellets were lysed with RIPA buffer and the cell lysates were subjected to Western blot analysis to detect to p53 and HIV-1 p24 (A) and NFAT, SP-1, AP-1, c-Fos and NF-ĸB (C). View Figure 1
It is well known that the HIV-1 LTR has recognition sequences for many cellular transcription factors, including NFAT, NF-κB, AP-1 and SP-1, which play very important roles in HIV-1 replication [7-9]. In order to examine whether p53 expression in increased activation of HIV-1 replication affects these host transcription factors, proteins from cell pellets were subjected to Western blotting to detect Ap-1, c-Fos, Sp-1, NFAT and NF-κB p65. As shown in Figure 1C, p53 expression dramatically upregulated expression of Ap-1, c-Fos, Sp-1, NFAT and NF-κB p65.
These data indicate that p53 can activate HIV-1 replication from HIV-1 latent infection by increasing expression of host transcription factors.
HMBA, originally developed as a potent inducer of some tumor cells [16], has been reported to activate the positive transcription elongation factor P-TEFb [11]. U1 cells were treated with HBMA with or without infection of adenovirus containing p53 or LacZ, for 3 days. HBMA treatment was found to activate HIV-1 replication and p53 expression with the presence of HMBA increased HIV-1 RNA production very significantly compared to Lac Z controls with presence of HBMA treatment (Figure 2A). These results suggest that p53 may have similar function as HBMA activities in activating HIV-1 replication from its latent state.
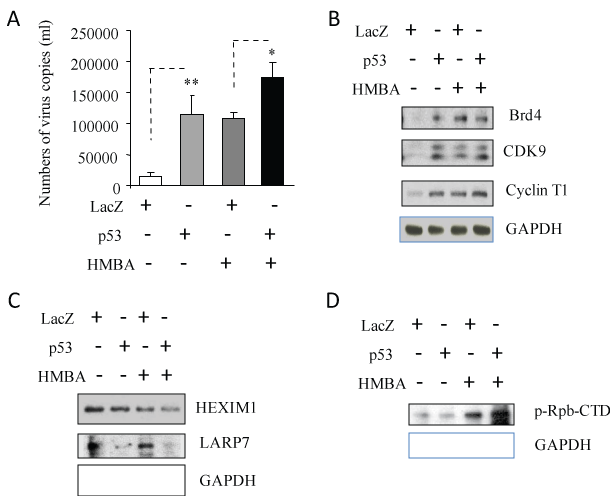 Figure 2: p53 expression could activate signaling pathways of positive transcription elongation factor b (P-TEFb). U1 cells were infected with adenovirus inserted a gene, p53 or LacZ for 2 hours, washed with PBS, and then cultured with the presence/absence of HMBA indicated for 3 days. After incubation for 3 days, (A) 140 µl of culture supernatants containing HIV-1 particles were used to isolate viral RNA. 10 µl in 50 µl of the RNA was used as template to perform real-time PCR. Known concentrations of HIV-1 (MN) viral RNA (serially diluted: 108 to 100 copies) were used as templates and quantitative RT-PCR performed to generate a standard curve. Each value represents the average concentration of six reactions in triple isolated repeats based on the standard curve. The value of p < 0.05 (*) was considered significant, p < 0.01 (**) very significant. The cell pellets were lysed with RIPA buffer and the cell lysates were subjected to Western blot analysis to detect to (B) Bed4, CDK9 and Cyclin T1; (C) HEXIM1 and LARP7; and (D) p-Rpb-CTD. View Figure 2
Figure 2: p53 expression could activate signaling pathways of positive transcription elongation factor b (P-TEFb). U1 cells were infected with adenovirus inserted a gene, p53 or LacZ for 2 hours, washed with PBS, and then cultured with the presence/absence of HMBA indicated for 3 days. After incubation for 3 days, (A) 140 µl of culture supernatants containing HIV-1 particles were used to isolate viral RNA. 10 µl in 50 µl of the RNA was used as template to perform real-time PCR. Known concentrations of HIV-1 (MN) viral RNA (serially diluted: 108 to 100 copies) were used as templates and quantitative RT-PCR performed to generate a standard curve. Each value represents the average concentration of six reactions in triple isolated repeats based on the standard curve. The value of p < 0.05 (*) was considered significant, p < 0.01 (**) very significant. The cell pellets were lysed with RIPA buffer and the cell lysates were subjected to Western blot analysis to detect to (B) Bed4, CDK9 and Cyclin T1; (C) HEXIM1 and LARP7; and (D) p-Rpb-CTD. View Figure 2
HMBA activates P-TEFb and its recruitment factor, bromodomain containing protein Brd4, thereby resulting in the activation of transcriptional elongation of HIV-1 [16] with formation of the P-TEFb complex including CDK9 and Cyclin T1 [11]. To verify whether p53 expression in reactivation of HIV-1 replication is involved in P-TEFb signaling pathways, cell lysates from Figure 2A were subjected to Western Blotting analysis to detect Brd4, CDK9 and Cyclin T1. As shown in Figure 2B, HBMA treatment and p53 expression could increase the expression of protein Brd4, CDK9 and Cyclin T1, suggesting that p53 expression is able to induce P-TEFb signaling pathway in reactivating HIV-1 replication.
The 7SK small nuclear Ribo Nucleo Protein (snRNP) plays a central role in RNA polymerase II elongation control by regulating the availability of active P-TEFb. In the inactive state, HEXIM1 (Hexamethylene bisacetamide inducible protein 1) and La Related Protein LARP7 interacts with P-TEFb through 7SK small nuclear ribonucleoprotein, resulting in inhibition of P-TEFb signaling pathways [17]. Both p53 expression and HMBA treatment downregulated expression of protein HEXIM1 and LARP7 (Figure 2C).
Transcription of many viruses is dependent on host cell factors such as RNA polymerase II (pol II) which is made up of 12 subunits. The Carboxyl-Terminal Domain (CTD) of the largest subunit of pol II plays a central role in transcriptional and co-transcriptional RNA processing. CTD modification generates a code that regulates interaction with transcription and RNA processing factors. Among the CTD modifications, the phosphorylation of Ser2 (Ser2P) of pol II CTD, is catalyzed by CDK 9 subunit of P-TEFb [11,16,17]. We found that HMBA treatment and increased expression of p53 expression resulted in increased phosphorylated pol II CTD (Figure 2D).
These results indicate that p53 is able to induce P-TEFb signaling pathways through downregulation of proteins, HEXIM1 and LARP7, which impede P-TEFb-signaling, upregulation of proteins, Brd4, CDK9 and Cyclin T1, which induce the elongation of HIV-1 transcription, and activation of pol II CTD, which plays the role directly in HIV-1 transcription and elongation in HIV-1 infected macrophages.
It has been reported that the Phosphatidyl Inositol 3-kinase (PI3K)/protein kinase B (Akt) signaling pathway is involved in the maintenance of HIV-1 latency, inhibition of the pathway can reactivate HIV-1 replication from its latent infection [18]. Figure 3A showed that p53 expression decreased the expression of protein, PI3K and Akt, suggesting that p53 affects PI3K/Akt signaling in U1 cells.
The Mitogen-Activated Protein Kinases (MAPKs) ERK1 and ERK2 (also known as p44/42 MAPK) are central components of signal transduction pathways activated by diverse extracellular stimuli. Previously we found that HIV super infection could activate ERK1/2 signaling in reactivation of HIV-1 latent infection in U1 cells [19]. To examine the effect of p53 expression on ERK1/2 activation and p38, U1 cells were infected with adenovirus containing p53 or LacZ for 3 days. As shown in Figure 3B, p53 expression displayed increased ERK1/2 phosphorylation and decreased p38 expression relative to LacZ controls.
 Figure 3: p53 expression can induce PI3K/Akt and MAPK signaling. U1 cells were infected with adenovirus inserted a gene, p53 or LacZ for 2 hours, washed with PBS and then cultured for 3 days. The cell pellets were lysed with RIPA buffer and the cell lysates were subjected to Western blot analysis to detect to (A) PI3K and Akt; (B) p38α/β and Erk1/2. View Figure 3
Figure 3: p53 expression can induce PI3K/Akt and MAPK signaling. U1 cells were infected with adenovirus inserted a gene, p53 or LacZ for 2 hours, washed with PBS and then cultured for 3 days. The cell pellets were lysed with RIPA buffer and the cell lysates were subjected to Western blot analysis to detect to (A) PI3K and Akt; (B) p38α/β and Erk1/2. View Figure 3
 Figure 4: p53 expression increased expression of CD4 and LEDGF/p75. U1 cells were infected with adenovirus inserted a gene, p53 or LacZ for 2 hours, washed with PBS, and then cultured for 3 days. The cell pellets were lysed with RIPA buffer and the cell lysates were subjected to Western blot analysis to detect to CD4 (A); and LEDGF/p75 (B).
View Figure 4
Figure 4: p53 expression increased expression of CD4 and LEDGF/p75. U1 cells were infected with adenovirus inserted a gene, p53 or LacZ for 2 hours, washed with PBS, and then cultured for 3 days. The cell pellets were lysed with RIPA buffer and the cell lysates were subjected to Western blot analysis to detect to CD4 (A); and LEDGF/p75 (B).
View Figure 4
Human macrophages are one of the main targets for HIV-1 infection, despite the moderately low surface expression levels of the main HIV-1 receptor, CD4 [20]. It was reported that HIV super infection upregulated CD4 expression and HIV-1 super infection promoted CD4 migration to lipid rafts in U1 cells [19]. Here, we found that p53 expression dramatically increased CD4 expression relative to LacZ control in U1 cells (Figure 4A).
The cellular transcriptional co-activator Lens Epithelium Derived Growth Factor (LEDGF)/p75 (p75) is an essential HIV integration cofactor and has gained prominence as a host factor involved in HIV integration [21]. Figure 4B showed that p53 expression in U1 cells induced higher levels of p75 protein expression relative to LacZ control.
These data indicate that p53 facilitates HIV-1 binding to host cells through increased expression of CD4 and integration into host chromosome through upregulation of integration cofactor p75.
It has been reported that the host-derived factor, COMM Domain-containing protein 1 (COMMD1), is able to reinforce HIV-1 latent infection through IκB-α stabilization in U1 cells, which inhibits HIV-1 replication by blocking NF-κB p65 activities in initiation of HIV-1 transcription [22]. To investigate whether p53 had an effect on expression of COMMD1 protein, U1 cells were infected with adenovirus containing p53 or LacZ for 3 days. Figure 5A shows that COMMD1 protein was downregulated by p53 expression relative to LacZ controls, suggesting that p53 can inhibit COMMD1 expression in U1 cells.
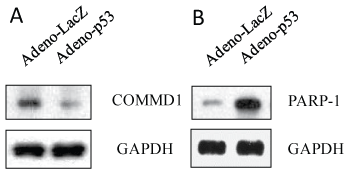 Figure 5: p53 expression suppressed COMMD1 expression and promoted PARP-1 expression. U1 cells were infected with adenovirus inserted a gene, p53 or LacZ for 2 hours, washed with PBS and then cultured for 3 days. The cell pellets were lysed with RIPA buffer and the cell lysates were subjected to Western blot analysis to detect to COMMD1 (A); and PARP-1 (B). View Figure 5
Figure 5: p53 expression suppressed COMMD1 expression and promoted PARP-1 expression. U1 cells were infected with adenovirus inserted a gene, p53 or LacZ for 2 hours, washed with PBS and then cultured for 3 days. The cell pellets were lysed with RIPA buffer and the cell lysates were subjected to Western blot analysis to detect to COMMD1 (A); and PARP-1 (B). View Figure 5
 Figure 6: p53 expression affected epigenetic regulators. U1 cells were infected with adenovirus inserted a gene, p53 or LacZ for 2 hours, washed with PBS, and then cultured for 3 days. The cell pellets were lysed with RIPA buffer and the cell lysates were subjected to Western blot analysis to detect to PCAF (A); HDAC1, HDAC2 and SIRT1 (B); and EZH2 (C).
View Figure 6
Figure 6: p53 expression affected epigenetic regulators. U1 cells were infected with adenovirus inserted a gene, p53 or LacZ for 2 hours, washed with PBS, and then cultured for 3 days. The cell pellets were lysed with RIPA buffer and the cell lysates were subjected to Western blot analysis to detect to PCAF (A); HDAC1, HDAC2 and SIRT1 (B); and EZH2 (C).
View Figure 6
Poly (ADP-Ribose) Polymerase 1 (PARP-1) is an abundant nuclear enzyme that catalyzes the successive transfer of the ADP-ribose moiety of NAD+ to a variety of nuclear proteins, including itself [23]. PARP-1 is required for the activation of NF-κB-dependent target genes, including HIV-1 LTR in reporter constructs and it may also play a role in HIV-1 integration [24]. p53 expression increased protein expression of PARP-1 compared to LacZ controls for 3 days (Figure 5B).
These data indicate that p53 affects the important host transcription factor, NF-κB, at different levels.
Cellular epigenetic factors can regulate HIV latency and reactivation by affecting the chromatin state in the vicinity of the viral promoter located to the 5' Long Terminal Repeat (LTR) sequence [25]. In turn, distinct HIV proteins can modulate the epigenotype and gene expression pattern of the host cells. Histone modifications via methylation and acetylation are well-studied post-translational protein modifications involved in regulating HIV-1 latency [25]. These modifications at a particular residue of histone tails can alter accessibility of the transcription factors, viral and RNA polymerizing machinery to the HIV-1 5' LTR [26]. Histone reversible acetylation is controlled by the activity of Histone Acetyl Transferases (HATs) and Histone Deacetylases (HDACs) that regulate transcription by selectively acetylating or deacetylating the ε-amino group of lysine residues in histone tails [2,27]. Generally, chromatin acetylation by HATs promotes chromatin opening and is associated with active euchromatin, whereas deacetylation by HDACs diminishes the accessibility of the nucleosomal DNA to transcription factors [2,28].
P300/CBP-Associated Factor (PCAF), also known as K (lysine) Acetyl Transferase 2B (KAT2B), is a human gene and transcriptional co-activator associated with p53 [29]. Several domains of PCAF can act independently or in unison to enable its functions. PCAF has separate acetyl transferase and E3 ubiquitin ligase domains as well as a bromodomain for interaction with other proteins. PCAF also possesses sites for its own acetylation and ubiquitination [29]. U1 cells infected with adenovirus containing p53 showed dramatic increase in PCAF expression relative to LacZ control (Figure 6A).
SIRT1 ((silent mating type information regulation 2 homolog) 1) is an enzyme that deacetylates proteins that contribute to cellular regulation. The HIV-1 Tat protein is a substrate for the deacetylase activity of SIRT1 which recycles Tat to its un-acetylated form, catalyzing a fundamental step to start new cycles of viral transcription [30]. p53 expression significantly decreased the expression of proteins, HDAC1, HDAC2 and SIRT1 (Figure 6B), resulting in activation of chromatin.
Among the histone modifications associated with gene silencing, much has been learned recently about the enzymes responsible for methylation of histone lysine residues. EZH2 is the catalytic subunit of Polycomb Repressive Complex 2 (PRC2), which is a highly conserved histone methyl transferase that targets lysine-27 of histone H3 [31]. p53 expression significantly suppressed EZH2 expression (Figure 6C).
These results indicate that p53 can modulate epigenetic regulators, including methylation of histones and upregulation of acetylation and inhibition of deacetylation, all of which play a role in reactivation of HIV-1 replication in HIV-1 latent infection.
p53 is a gene that codes for a protein that regulates the cell cycle and hence functions as a tumor suppressor gene. Cellular stresses, such as DNA damage, cause stabilization and activation of the p53 tumor suppressor protein [5]. The genes regulated by p53 include genes encoding cell surface proteins, mitochondrial proteins and cytoplasmic proteins. The p53-mediated transcriptional response to DNA damage is extremely complex. P53-regulated gene expression patterns differ not only in different cell types but also in response to different induction signals, resulting in one of two different outcomes including cell cycle arrest or apoptotic cell death [5]. Cell death induced by p53 is executed by the caspase proteases, which, by cleaving their substrates, lead to the characteristic apoptotic phenotype. Caspase activation by p53 results in the release of apoptogenic factors from the mitochondria [5]. Infection with HIV results in the progressive destruction of CD4 T lymphocytes as well as other immune responder cells [32]. The depletion of CD4 lymphocytes leads to subsequent immune dysfunction responsible for the occurrence of opportunistic infections associated with AIDS [1]. Programmed cell death, or apoptosis, has been shown to be a mechanism underlying the depletion of CD4 lymphocytes in HIV-infected individuals [1]. In 1996, it was reported that HIV-1 infection could activate or up regulate p53 protein, which stimulated Bax activation downstream [33]. We also found that both HIV-1 and HIV-2 infection could induce apoptotic cell death through the p53/Bax signaling pathway and HIV-1 infection increased p53 expression to higher levels than HIV-2 infection [15].
Previously, we reported that expression of proapoptotic proteins, including Fas Ligand (FasL), FADD, or p53 significantly increased HIV-1 virus production. In contrast, the expression of antiapoptotic molecules, such as FLIP, Bcl-XL and XIAP, decreased HIV-1 virus production. Knockdown of Bax with siRNA and FADD with expression of its antisense mRNA also inhibited viral replication and the caspase-3 inhibitor, Z-DEVD and decreased virus production [13]. These results indicate that proapoptotic proteins can increase HIV-1 replication and antiapoptotic molecules are able to inhibit HIV-1 replication, suggesting that HIV-1 has evolved ways to inhibit host cell apoptosis [12], an important way for the virus to enhance its replication when host cells initiate the apoptotic program as a way of limiting replication within the host. We and other investigators show that, in addition to HIV, infection with influenza virus (H1N1) [14], HSV-1 [34], KSHV [34], HHV6A, HHV6B, HHV7 and EBV [35] causes the host cell to undergo apoptosis and they adopt an emergency escape mechanism that leads to the rapid production of large amounts of virus. It has been shown that p53 expression is sufficient to initiate the emergency escape process [13].
Thus far, effective approaches to access and deplete the long-lived reservoir of cells latently infected with HIV-1 remain an elusive goal, and the best strategy to block reappearance of virus after the anti-retrovirus therapy is discontinued has not been identified. Recently, it has been reported that HIV-1 super infection [19], co-infection with influenza virus (H1N1) [36] and apoptosis [12] can induce activation of HIV-1 in latently infected cell lines. p53 expression alone can be sufficient to activate HIV-1 replication in U1 cells (Figure 1). HIV-1 gene expression is regulated mainly by activation of the LTR after integration into the host genome. As inducible trans activators of HIV-1, HIV-1-derived transcription factor Tat and host-derived transcription factor NF-κB, NFAT, AP-1 and SP1 directly bind to the HIV-1 LTR and trans activate HIV-1 gene expression by forming the transcriptional initiation HIV-1 complex [7-9,19]. p53 expression also activates or enhances these host transcription factors (Figure 1).
Activation of latent HIV-1 infection occurs at the level of elongation of transcription and requires HIV-1 Tat binding to the Transactivation response element at the start of HIV-1 transcripts [8]. The CDK9/cyclin T1 complex is a key factor for productive elongation of transcription of HIV-1 genomes through phosphorylation of the Carboxyl-Terminal Domain (CTD) of the large subunit of RNA polymerase II [11] and the inhibition of CDK9 impairs HIV-1 replication [37]. CDK9 directly interacts with the C-terminal domain (aa361-393) of p53 and p53 interacts with the N-terminal domain of CDK9, in which CDK9 phosphorylates serine 392 of p53 through their direct physical interaction [38,39], while p53 binds and activates the CDK9 promoter [40]. These observations are consistent with our findings that p53 expression can activate P-TEFb signaling pathways (Figure 2).
The transcription factor NF-κB is important for HIV-1 transcription initiation in primary HIV-1 infection [36] and reactivation in latently HIV-1-infected cells [19]. It was reported that low levels of nuclear NF-κB in resting CD4+ T cells may support the establishment of HIV latency [41]. Upon cell activation, the p50/p65 heterodimer binds to NF-κB sites in the HIV LTR, recruits Histone Acetyl Transferase (HAT) to acetylate the histone tails, opens nucleosomes to facilitate HIV transcription and recruits HDAC1 leading to the suppression of HIV expression [42]. Our data showed that, in addition to activating NF-κB p65 itself, p53 expression assists and is involved in NF-κB p65-driven reactivation of HIV-1 latently infected U1 cells. An increased expression of PARP is required for the activation of NF-κB-dependent target genes, including HIV-1 LTR in reporter constructs; a decreased expression of COMMD1 reinforces HIV-1 latent infection through IκBα stabilization [22].
The state of chromatin (the packaging of DNA in eukaryotes) has long been recognized to have major effects on levels of gene expression, and numerous chromatin-altering strategies including histone modification are employed in the cell to bring about transcriptional regulation [1]. Histone acetylation is high in active chromatin. Inhibition of acetyl transferases including P300 and PCAF inhibits cell proliferation [29], downregulates the expression of p53 downstream genes, and induces cell apoptosis with increased levels of activated caspase-3 and caspase-9 and decreased mitochondrial membrane potential in some cancer cells [29]. Histone Deacetylation (HDAC) represses chromatin. SIRT1 represses p53 function via deacetylation, promoting tumor growth in carcinoma cells [30] and SIRT1 inhibition increases p53 acetylation, and Bax expression and induces caspase 3 cleavages. Here, we found that p53 expression could increase acetyl transferase PCAF expression, and downregulate proteins HDAC1/2 and SIRT1 in U1 cells. The mechanism needs further investigation.
Inhibition of p53 signaling by Pifithrin-α, an inhibitor of p53 transcriptional activity, was reported to be resistant to reactivation of latent HIV-1 infection by αCD3/αCD28-coated magnetic beads in central memory CD4 T cells [43]. The results are consistent with our conclusion that inhibition of proapoptotic proteins can decrease HIV-1 replication and reactivation of latent HIV-1 infection. However it was reported that p53 silencing significantly enhanced HIV-1 replication in infected cells [44]. In studies of influenza virus co-infection [36,45], a biphasic pattern of p53 was found with an early phase of infection and apoptotic phase [45]. p53 significantly enhances virus replication only in the late apoptotic phase with a large amount of p53 protein expression [14]. Further investigation is needed on whether the biphasic pattern of p53 affects HIV-1 replication in primary infection and reactivation of replication from latent HIV-1 infection. In HIV super infection of latently infected U1 cells, we found that super infection with HIV-1 displayed a very significantly activated HIV-1 replication with a large amount of p53 protein expression compared to HIV-2 super infection after 3 days of infection [19].
In conclusion, the monocytic U1 cell line, which shows minimal constitutive expression of HIV-1, when infected with adenovirus containing p53 or LacZ, displayed an increase in HIV-1 replication. p53 expression reactivated HIV-1 replication from its latent infection through increased expression of host transcription factors, NF-κB, NFAT, AP1, SP1 and c-Fos, and inducing P-TEFb signaling pathways. p53 expression upregulated PARP-1 and downregulated COMMD1 to increase NF-κB activity. P53 also affected epigenetic factors with increased histone acetylation, decreased HDAC1/2 and SIRT1 and histone methylation to open nucleosomes to facilitate HIV transcription.