CTGF/CCN2 is involved in many cellular events related to proliferation and migration. Its main physiological role is related to the promotion of endochondral ossification, besides regeneration, protection of bone and articular cartilage, and angiogenesis. Skeletogenesis, the process by which bone and articular elements are formed, comprises endochondral and intramembranous ossification. However, the signaling pathways that mediate CTGF/CCN2 expression in chondrocytes have not been fully understood. Osteoarthritis, defined by synovial inflammation, hyaline cartilage degeneration and subchondral bone thickening that affects the whole synovial joint. CTGF/CCN2 is expressed by hypertrophic chondrocytes of chondro-osteophytes, resembling the normal endochondral ossification appearance. Hence, the goal of this paper is to review the role of CTGF/CCN2 in skeletogenesis, focusing on cartilage repair and the development of OA.
CCN2, CTGF, Musculoskeletal system, Bone healing, Chondrocyte
ATP: Adenosine Triphosphate; BMP: Bone Morphogenetic Proteins; CCL: Chemokine Ligand 2; CCN2: Cellular Communication Network 2; CD: Cluster of Differentiation; CT: C Terminal domain; CTGF: Connective Tissue Growth Factor; ECM: Extracellular Matrix; FGF: Fibroblast Growth Factor; FLS: Fibroblast-Like Synoviocytes; HCS: High Content Screening; IGFBP: Insulin-Like Growth Factor Binding Protein; IL: Interleukin; MMP: Metalloproteinases; MSC: Mesenchymal Stem Cells; NFκB: Nuclear Factor Kappa B; OA: Osteoarthritis; SOX: SRY-related HMG-box genes; SP: Signal Peptide; TGF: Tumor Growth Factor; TSP1: Thrombospondin-1; VEGF: Vascular Endothelial Growth Factor; VWC: Von Willebrand type C domain
Connective tissue growth factor (CTGF/CCN2) was described in 1991 during experiments involving human endothelial cells [1]. Its name arose from its mitogenic and chemotactic properties in fibroblasts [2], although it is also produced by many cell types as osteoblasts [3] and chondrocytes [4]. CTGF/CCN2 is a member of the CCN family (CYR61/CTGF/NOV) [5-7]. This family unified in nomenclature a group of proteins, which shared a multimodular structure (Figure 1) with a N-terminal secretory signal followed by four conserved modules with homologies to insulin-like growth factor binding proteins, von Willebrand factor type C repeat, thrombospondin type I repeat and a carboxy-terminal domain containing a cysteine knot [6,7].
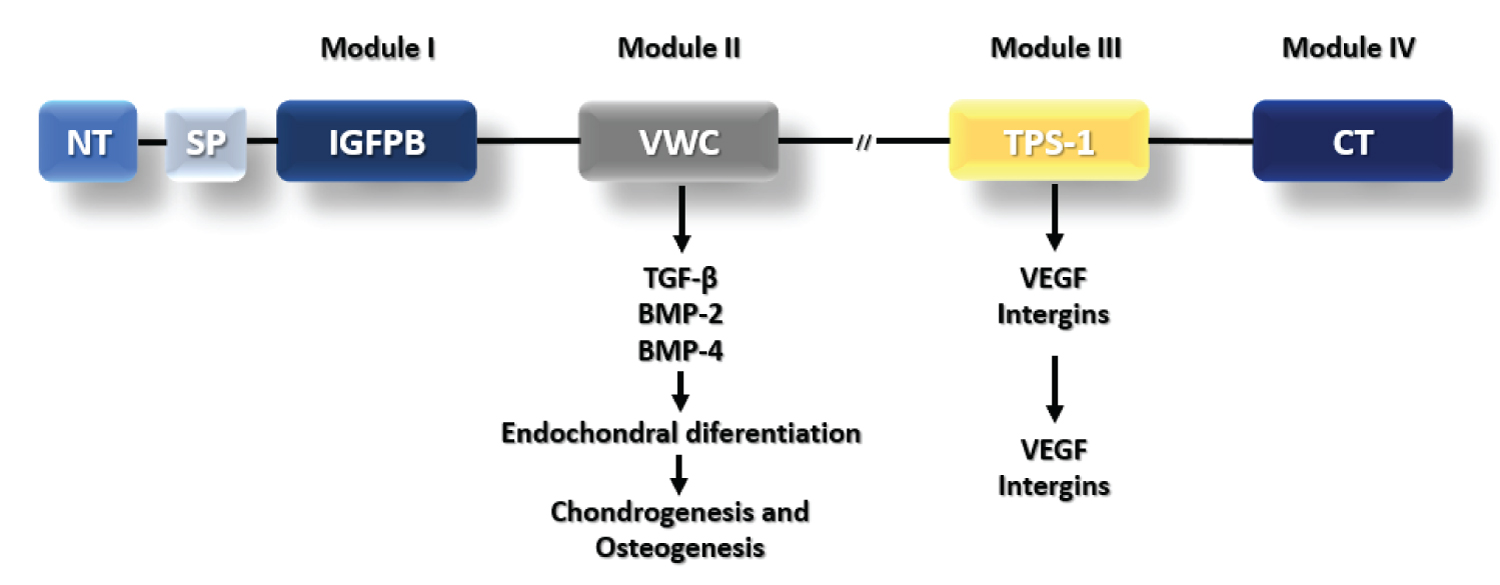 Figure 1: CTGF Structural domains and functions. CTGF protein has a N-terminal followed by signal peptide (SP) and more four domains. Module I is an insulin-like growth factor (IGF)-binding domain. Module II is a Von Willebrand type C domain that interacts with TGF-beta, BMP-2 and BMP-4 for skeletogenesis. Module III is a Thrombospondin-1 domain that interacts with VEGF and Integrins for angiogenesis. Module IV is a C-terminal domain. The symbol -//- represents cysteine rich domains that region is susceptible to enzymatic cleavage.
View Figure 1
Figure 1: CTGF Structural domains and functions. CTGF protein has a N-terminal followed by signal peptide (SP) and more four domains. Module I is an insulin-like growth factor (IGF)-binding domain. Module II is a Von Willebrand type C domain that interacts with TGF-beta, BMP-2 and BMP-4 for skeletogenesis. Module III is a Thrombospondin-1 domain that interacts with VEGF and Integrins for angiogenesis. Module IV is a C-terminal domain. The symbol -//- represents cysteine rich domains that region is susceptible to enzymatic cleavage.
View Figure 1
CTGF/CCN2 is involved in many cellular events related to proliferation [1] and migration [8]. Recently, its role in cell migration related to metastasis was reviewed [6]. However, CTGF/CCN2 role is far beyond, since its main physiological role was indicated to be the promotion of endochondral ossification, besides additional roles in regeneration, protection of bone and articular cartilage, and angiogenesis [9]. Many papers reviewed the participation of CTGF/CCN2 in skeletogenesis [5,10-12]. In this aspect, CTGF plays an important role in skeletal development, and a secondary one in cell proliferation [13,14]. Furthermore, it promotes in vitro differentiation of growth and articular cartilage from rabbits, depending on cell type, towards to endochondral ossification or formation of articular cartilage [15].
There is evidence that CTGF/CCN2 is important in the growth plate for maintaining extracellular matrix (ECM) and chondrocyte survival [16]. Despite this, CTGF/CCN2 overexpression triggers pathways leading to fibrosis, due to excess collagen deposition in organs [16,17]. The ability of inducing synovial fibrosis and cartilage damage by CTGF/CCN2, hallmarks of osteoarthritis (OA), have also been demonstrated [18]. Interestingly, some studies showed that OA follows a similar way to endochondral ossification, as chondrocytes leave the stable phenotype and continue to hypertrophy, towards terminal differentiation [19]. Hence our interest in discussing CTGF/CCN2 involvement in both processes. The goal of this paper is to review the role of CTGF/CCN2 in skeletogenesis, focusing on cartilage repair and the development of OA.
Skeletogenesis, the process by which bone and articular elements are formed, comprises endochondral and intramembranous ossification. Endochondral ossification responds for most of the skeletal elements and is divided into two steps. The first involves the aggregation of mesenchymal cells in sites where the skeletal elements will be formed. The latter corresponds to the differentiation of three different cell types through cell proliferation and differentiation events [20]. Endochondral bone formation begins with the condensation of mesenchymal cells, which differentiate into chondrocytes, secreting a matrix rich in type II collagen and forming a cartilage template. Chondrocyte proliferation and matrix production lead to cartilage enlargement. Then, chondrocytes in the center of the template stop to proliferate, hypertrophy, and change their phenotype into producing type X collagen. Hypertrophic chondrocytes attract blood vessels through the production of vascular endothelial growth factors (VEGF), directing adjacent perichondrial cells to become osteoblasts, which form a bone collar [21]. Subsequently, hypertrophic chondrocytes undergo apoptosis, and the matrix left behind provides a scaffold for osteoblasts invasion along with blood vessels [21]. While the cells in the center of the template enlarge to differentiate into hypertrophic chondrocytes, as described in the paragraph above, cells of the periphery start to proliferate in an organized manner, in parallel to the longitudinal axis of the bone, originating the growth plate [22].
Many signaling pathways are involved in skeletogenesis (Figure 2), such as fibroblast growth factors (FGF), bone morphogenetic proteins (BMP), SOX9, Runx2 [21,23] and Smad [23]. However, the signaling pathways that mediate CTGF/CCN2 expression in chondrocytes has not been fully understood. CTGF/CCN2 regulates chondrocyte proliferation, ECM synthesis and angiogenesis, being important in many steps of chondrogenesis [24]. In addition, it has an important role in VEGF localization in hypertrophic chondrocytes [24], which is essential for growth plate angiogenesis [25]. VEGF expression can be induced by CTGF/CCN2 directly binding to transforming growth factor β (TGF-β), enhancing TGF-β ability to interact with its receptors [26]. CTGF/CCN2 also regulates VEGF activity [24], stimulating vascular endothelial cells proliferation, migration, and tube formation, acting as a paracrine factor released by hypertrophic chondrocytes during cartilage replacement by bone [13].
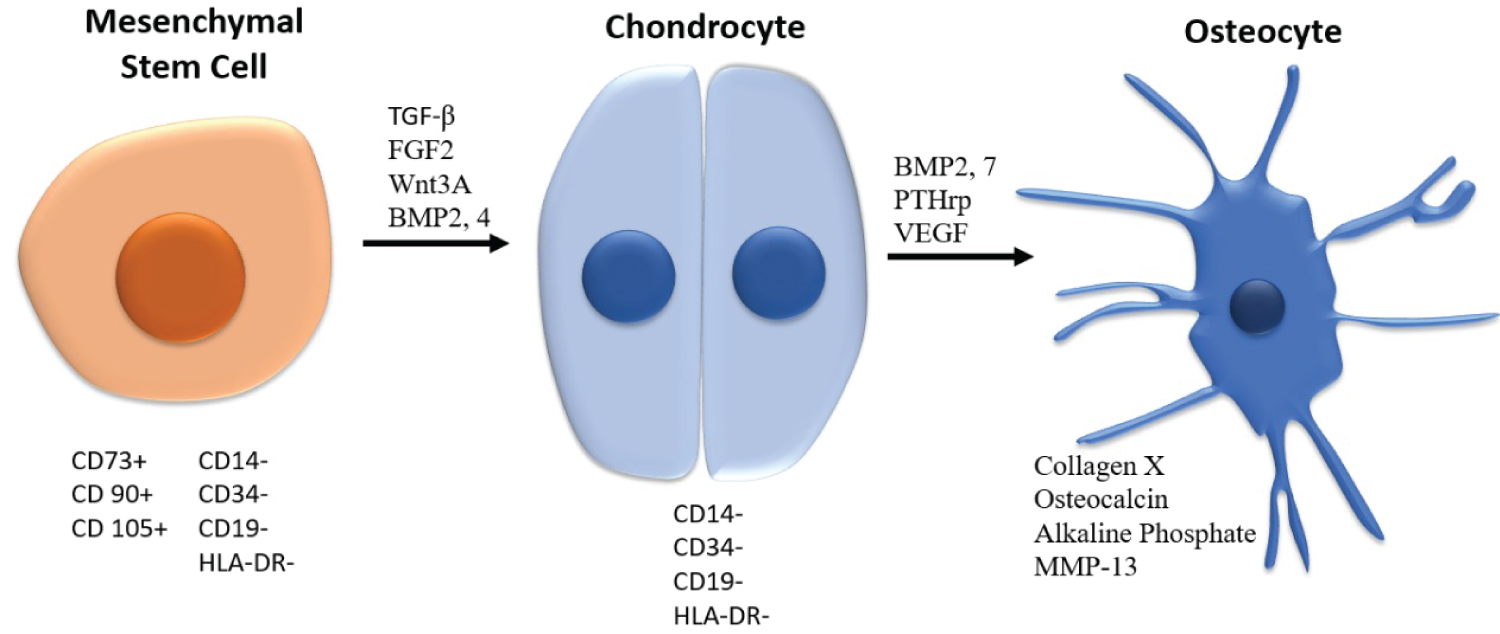 Figure 2: Main cellular types and pathways involved in skeletogenesis. Mesenchymal stem cells, chondrocytes and osteocytes are the main cell types involved in skeletogenesis, and present specific surface markers.
View Figure 2
Figure 2: Main cellular types and pathways involved in skeletogenesis. Mesenchymal stem cells, chondrocytes and osteocytes are the main cell types involved in skeletogenesis, and present specific surface markers.
View Figure 2
Another way by which CTGF/CCN2 can act during skeletal development is through binding to BMP and TGF-β, inhibiting the first and enhancing the latter [24]. TGF-β, which regulates cell proliferation and differentiation in many biological processes [27], including tissue repair and inflammation [28], stimulates the expression of CTGF/CCN2 in hypertrophic chondrocytes, promoting both proliferation and differentiation of proliferating and mature chondrocytes, respectively, in a paracrine manner [13]. Although the mechanism by which CTGF influences TGF-β is not clear, there is evidence indicating a synergy between both cytokines [29]. Besides, CTGF and TGF-β seem to have a role as chondroprotective agents in articular cartilage [9,30]. TGF-β canonical signaling, transduced via Smad 2 and Smad 3 [31], promotes early stages of chondrocyte differentiation and maturation [32]. It has also been reported that both physiological and excessive mechanical compression activate Smad2/3P signaling, which protects articular cartilage and blocks chondrocyte terminal differentiation [33]. On the other hand, aged cartilage has a decreased ability for mechanically induced Smad2/3P signaling activation [34]. Recently, it was described that CTGF depletion leads to a paradoxical increase in Smad2 phosphorylation which resulted in thicker and protected from OA cartilage [29]. Rac1 signaling and actin organization in chondrocytes also regulates CTGF/CCN2 expression, indicating the participation of the cytoskeleton in its regulation. CTGF/CCN2 gene response to Rac1 signaling is mediated by the Smad binding site in the CTGF/CCN2 promoter [27]. CTGF/CCN2 interacts with BMP-2 and the complex formed regulates the differentiation of chondrocytes [35]. BMP-2 and insulin-like growth factor 1 (IGF-1) induce CTGF/CCN2 expression, suggesting that they are involved in bone remodeling through induction of CTGF/CCN2, and, hence, indicating that CTGF/CCN2 is important in bone remodeling and in endochondral ossification [14].
Articular cartilage is the tissue responsible for providing a low shear stress and frictionless surface for joint movement [36]. Chondrocyte, the only type of cell in adult human cartilage, represents 1 to 2% of tissue volume [37,38]. Type II collagen is the main component of cartilage ECM [36,39]. Chondrocytes and matrix interdependence maintain tissue viability since the ECM protects cells from mechanical damage [37]. Water contributes with 80% of cartilage weight and interacts with matrix macromolecules influencing on the tissue biomechanical properties [36]. ECM transduces signals from the microenvironment, regulating the release of growth factors. ECM remodeling depends on balancing the production and degradation of specific ECM components. Metaloproteinases (MMP) plays a role in this process, cleaving growth factors and their binding proteins, and so inhibiting or activating specific signaling pathways. MMP-1, MMP-2 and MMP-3 have increased expression in fibroblasts due to CTGF/CCN2 overexpression, suggesting CTGF/CCN2 controls ECM production and degradation [24]. Ctgf/Ccn2-deficient mice show skeletal dysmorphism associated to decreased chondrocyte proliferation, delayed differentiation, impaired ECM composition and insufficient vascular invasion. This phenotype results in sever chondrodysplasia, leading to death at birth because of respiratory failure. In Ctgf/Ccn2-deficient mice, cartilage has inferior mechanical properties, turning it susceptible to deformation and distortion during development. Ctgf/Ccn2 seems to be required to regulate chondrocyte proliferation at later stages of chondrogenesis and not at early ones [24]. Deformed cartilage seen in mutants suggested CTGF/CCN2 is required for synthesizing normal ECM components levels. But mutant neonates showed no differences in types II and X collagen distribution [24].
Overexpression of Ctgf/Ccn2 in mice, instead, protects articular chondrocytes against degenerative changes by induction of chondrocyte proliferation and proteoglycan synthesis [17,40]. The role of CTGF in protecting chondrocytes form stress is direct, not a consequence of defective growth plate angiogenesis, although it’s exact mechanism remains unclear [17]. In a study using two different animal models (mice and rat), it was shown that CTGF/CCN2 had the ability to repair both damaged and osteoarthritic articular cartilage. CTGF/CCN2 was strongly expressed in clustered chondrocytes, which represents a high proliferative response, while in normal cartilage expression was low, suggesting this factor plays an important role in articular cartilage repair [41]. Regeneration of cartilage is regulated by growth factors, such as TGF-β and BMP-2, which interact with CTGF/CCN2. In this same study, recombinant CTGF/CCN2 treatment of mesenchymal stem cells (MSC) presumably resulted in an increased production of type II collagen and aggrecans, specific of articular cartilage. The mechanism of MSCs regulation by CTGF/CCN2 was not clarified, but one possible explanation seems to be stimulating the condensation of MSCs, which initiates chondrogenesis [41]. Another study showed suggested that chondrocyte differentiation and hypertrophy during cartilage disease could stimulate CTGF/CCN2 production, which in turn acted as a paracrine and autocrine growth factor promoting proliferation and differentiation of the cells. CTGF/CCN2 was expressed by hypertrophic chondrocytes of chondro-osteophytes, resembling the normal endochondral ossification appearance. Its predominant expression might have been related to TGF-β high levels [42]. BMP-4 binding to CTGF/CCN2 can be partially completed by TGF-β1, suggesting CTGF/CCN2 may also bind to TGF-β1. Binding of BMP-4 to CTGF/CCN2 inhibits BMP signaling by avoiding BMP-4 binding to its cognate receptor. CTGF/CCN2 enhances TGF-β1 signaling by receptor and cell-surface binding [26]. CTGF/CCN2 could also act in the production of type I and II collagen or in the production of proteoglicans by chondrocytes, in the repair response [42]. A speculation is that increased CTGF/CCN2 expression by OA chondrocytes might have a role in the promotion of fibrosis in damaged cartilage regions [42].
Osteoarthritis, defined by synovial inflammation, hyaline cartilage degeneration and subchondral bone thickening that affects the whole synovial joint [43] is the main cause of disability worldwide. Primary osteoarthritis is age-related and characterized by the onset of joint pain and stiffness [44]. Some reviews suggested that hypertrophic-like changes are present in late stage OA [40] suggesting that endochondral ossification signals may be important for OA progression and indicating possible therapeutic targets for treatment [45]. A strong positive immune reaction to CTGF/CCN2 was seen in chondrocyte clusters, next to damaged cartilage surfaces and in hypertrophic chondrocytes in chondro-osteophytes, suggesting a relation between CTGF/CCN2 expression and cartilage degeneration and chondrocyte differentiation [42]. It was confirmed that CTGF/CCN2 concentrations were higher in synovial fluid of OA patients than in non-affected ones. IL-6 was identified as a target protein for the CTGF/CCN2 signaling pathway, regulating cell inflammatory response. CTGF/CCN2 induced inflammatory cytokines as IL-6 and enhanced the inflammatory response in OA [46]. Intracellular ATP level is maintained at higher levels in chondrocytes in the presence of CTGF/CCN2 than in its absence. Lack of CTGF/CCN2 during development resulted in increased ribosomal protein genes expression and decreased ECM and metabolism genes. In the growth plate, CTGF/CCN2 is produced by chondrocytes in the pre-hypertrophic layers in large amounts. So, ATP depletion could be considered as a causative factor in OA development [47].
CTGF/CCN2 promoter contains an NFκB responsive element which expression is activated during smooth muscle cells response to mechanical stretch. NFκB in growth plate may be regulated by CTGF/CCN2. In a chemically induced knee OA model, recombinant CTGF/CCN2 accelerated articular cartilage repair. This effect may be due to its modulation of NFκB. It is unknown if NFκB helps on chondrocyte survival. Besides pro-survival functions in chondrocytes, it promotes the expression of pro-inflammatory genes in OA chondrocytes [17]. Some results indicated that CTGF/CCN2 cooperates with IL-1β amplifying the inflammatory response, and leading to the production of cytokines, chemokines and MMPs in OA fibroblast-like synoviocytes (FLS). The data demonstrated also that CTGF/CCN2 synergistically enhanced IL-6 production and acted on OA FLS in vitro to enhance IL-8, CCL2 and CCL20 production. The augmented production of these mediators indicates that CTGF/CCN2 may be involved in the amplification of IL-1β induced inflammatory response in OA. These effects resulted in the production of catabolic mediators that lead to synovitis and articular destruction seen in OA [48].
In chondrocyte cultures, CTGF/CCN2 gene was easily and effectively transduced into articular chondrocytes using adenoviral vector. It stimulated chondrocyte maturation, without inducing hypertrophy. This strategy may be useful in the repair of damaged articular cartilage, since it stimulated chondrocyte proliferation, but not terminal hypertrophy and calcification. The increase in proteoglycan synthesis, a marker of chondrocyte maturation, was dose dependent. Both articular and growth plate chondrocyte phenotypes expression were stimulated, however, type X collagen expression was increased only in growth articular cells, while undetectable in the articular ones [15].
An association of in vivo and in vitro experiments involving auricular cartilage indicated that CTGF/CCN2 could be useful for the repair and reconstruction of elastic cartilage, given its ability of enhancing cell proliferation, proteoglycan synthesis, elastin, and type II collagen expression [30]. Recently, it has been demonstrated that each of the CTGF/CCN2 independent modules (IGFBP, VWC, TSP1 and CT) had strong effect in the expression level of both the aggrecan and type II collagen in HCS-2/8. Intact CTGF/CCN2 promotes proliferation and differentiation of chondrocytes partly through this signaling pathway. These independent modules can be more stably produced, purified, and stored than the intact CTGF/CCN2. Although further clinical investigation is necessary to evaluate its potential for therapeutic usage, among the single modules, TSP1 alone yielded stronger regenerative effects on damaged cartilage than intact CTGF/CCN2, representing a promising candidate for clinical use against OA [49]. Moreover, the development of clinical trials both for investigating CTGF-antagonists drugs in OA treatment and for the use of CTGF as OA diagnosis biomarker have been suggested [50].
CTGF is involved in many processes regarding cell proliferation and migration, including in skeletogenesis and cartilage repair, which are resembled in some stages of osteoarthritis development. Hence, CTGF emerges not only as a promising biomarker for OA diagnosis, but also as a therapeutic target for drug development.
Not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing interests.
Not applicable.
EBS and DPA: Extensive review of the literature, writing and final review of the paper.
Not applicable.