Over the past decade, there has been a rising interest in the use of carboranes as a potential pharmacophoric moiety in the development of new drugs for the treatment of various types of cancer. The unique physical and chemical properties of carboranes make their use attractive in drug development. In several instances, the inclusion of carboranes into a drug structure has increased the agent's binding affinity, potency, or bioavailability. The purpose of this review is to highlight applications of carboranes to the medicinal chemistry of cancer.
Cancer is the second leading cause of death in the United States, exceeded only by heart disease. It is estimated that 1,685,210 new diagnosis and 595,690 American deaths will be due to cancer this year [1]. The fight against cancer involving early detection and new methods of treatment are crucial. Since their initial discovery during the 1960's, researchers have utilized carboranes in the search for new and effective treatments for cancer [2,3]. Formally named dicarba-closo-dodecaboranes, carboranes containing two carbon atoms are icosahedral clusters containing carbon, boron, and hydrogen and exist as ortho 1, meta 2, and para 3 isomers, depending on the relative position of the two carbon atoms (Figure 1). Carboranes have a general formula of C2B10H12 and possess unique chemical and physical properties that suggest their potential use as a moiety for the design of new drug molecules with enhanced activities and/or target selectivies [3]. Some of these properties include high oxidative and thermal stability, hydrophobicity, and low toxicity [4]. Owing to their stability, carboranes also may increase the in vivo stability and bioavailability of pharmaceuticals that might otherwise rapidly metabolize [5]. Furthermore, selective chemical substitution of each carbon or boron atom in these clusters allows for their use as rigid, three dimensional scaffolds upon which to construct new drug molecules.
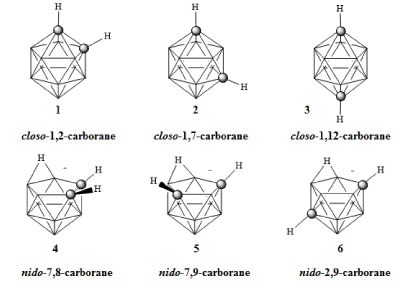 Figure 1: Ortho-(1), meta-(2), para-carborane (3), and nido-carboranes (4-6) [3]. View Figure 1
Figure 1: Ortho-(1), meta-(2), para-carborane (3), and nido-carboranes (4-6) [3]. View Figure 1
Nearly all past biomedical research involving carboranes has focused on their use in the design of boron delivery agents for boron neutron capture therapy (BNCT) [2]. This binary radiation therapy depends on the selective delivery of a high concentration of boron-10 atoms to targeted tissues. It is generally accepted that a minimum concentration of 30 ppm is required for successful BNCT therapy and this concentration is equivalent to the delivery of approximately 109 boron atoms to each targeted cell. As BNCT human clinical trials have been attempted nearly continuously for the past five decades, there is a large body of literature associated with the use of carboranes for the development of BNCT boron agents. There are several good reviews and books covering that work [6-13]. While BNCT requires the delivery of large quantities of boron to diseased tissue, (requiring concentrations of boron in the low millimolar range) there is an increasing interest to use carboranes to synthesize new and highly potent drugs which operate at the opposite extreme of the concentration range, namely nanomolar, to picomolar.
Ortho-carborane and its carbon-functionalized derivatives may be prepared by reacting acetylene (or functionalized acetylene) with the decaborane derivative B10H12L2, where L is a weak Lewis base [3,14]. The acetylenes used in these reactions include a wide range of functional groups such as esters, halides, carbamates, ethers, and nitro groups; however, such reactions are not successful in the presence of nucleophilic species such as alcohols, acids, or amines. Under an inert atmosphere, ortho-carborane will thermally isomerize to meta-carborane at temperatures ranging from 400-500 ℃ and to para-carborane at 600-700 ℃ [2]. The CH groups in the carboranes are weakly acidic and can be deprotonated using a strong base to produce nucleophiles [5]. The pKa values for ortho-, meta-, and para-carborane are 22.0, 25.6, and 26.8 respectively [15,16]. Separately, the boron vertices may be functionalized by reactive electrophiles. In turn, a wide range of carborane derivatives may be synthesized regioselectively without the need for complex protecting groups. Bases, such as pyrrolidine or fluoride, may be used to generate nido-carboranes 4, 5, and 6 (Figure 1) [17-21]. Such reactions convert the extremely hydrophobic closo-carboranes to the hydrophilic nido-carboranes [22-24].
Pharmacophore approaches involving various ligand-based and structure-based methods have evolved to be one of the most successful concepts in medicinal chemistry. A pharmacophore was defined in 1909 by Ehrlich as a molecular framework that carries the essential features responsible for a drug's biological activity [25]. Considerable progress has been made on pharmacophore technology in the past two decades and has made pharmacophore approaches a major tool in drug discovery. Recent developments have expanded the boron carrier approach of carboranes for BNCT to be used as frameworks in developing newly improved pharmacophores. Compared to aryl rings, the superior synthetic flexibility of carboranes allow them to be extremely useful in fine tuning the pharmacokinetics of newly developed drug candidates [5]. They are optimal for chemical modification by functionalizing the weakly acidic CH groups and boron atoms, and have the ability to employ various isomers (1-3) to optimize the binding affinity with other substituents [26]. The closo-carborane is air and moisture stable and is known to possess "superhydrophobicity". Their high partition coefficient values are known to surpass common bioisosteres such as aryl, cycloalkyl, and adamantyl groups [5]. Moreover, their high stability and low toxicity towards cells make them interesting compounds for therapeutic applications [2].
It has been apparent for close to a century that the steroid estrogen has played a major role in breast cancer [27]. When molecules, such as estrogen hormones, bind to the ligand binding domain of the estrogen receptor the protein forms a dimer. The dimerized form of the estrogen receptor binds to DNA promotor elements which results in the initiation of gene transcription [2,5]. Antagonism of the estrogen receptor is a mechanism used for the treatment of hormone responsive cancers [28-32]. The molecules depicted in Figure 2 represent the hormone 17β-estradiol 7 and the estrogen receptor alpha (ERα) antagonists tamoxifen 8 and 4-hydroxytamoxifen 9. Endo and coworkers synthesized a number of carborane containing estrogen receptor agonists 10a-g and antagonists 11-12 depicted in Figure 2 [28,29]. The activities of the estrogen agonists 10a-g were analyzed; the carborane derivative 10c exhibited at least 10-fold greater potency than 17β-estradiol 7 both in vitro and in vivo [29]. Remarkably, in comparison with the natural molecule, these carborane containing agonists are quite simple in both structure and synthesis, containing no carbon steriocenters. Based on the inhibition of ERα, derivative 12 demonstrated the greatest antiestrogen response, exhibiting an activity that was comparable to tamoxifen [28].
 Figure 2: Hormone and ERα antagonists (7-9), estrogen receptor agonist carborane derivatives (10a-g), and estrogen receptor antagonist carborane derivatives (11-12) [28]. View Figure 2
Figure 2: Hormone and ERα antagonists (7-9), estrogen receptor agonist carborane derivatives (10a-g), and estrogen receptor antagonist carborane derivatives (11-12) [28]. View Figure 2
The estrogenic response of these compounds in the uterine and bone tissues of ovariectomized mice was measured. The assays measured the inhibition of 17β-estradiol binding to the ERα receptor at concentrations of 1 × 10-9 M. The binding affinity for the ERα receptor for 7, 11, and 12 were 0.1 nM, 1.0 nM, and 60 nM respectively [28]. The most potent inhibition of the transcriptional response to 17β-estradiol was exhibited by 12, thus the authors noted that the antagonistic response does not directly correlate with the drugs binding affinity. They also noted that while inducing an agonistic effect on the estrogen receptor in the bone tissue of the mice, 11 exhibited antagonistic effects on the estrogen receptor in the uterine tissue. This observation supports the potential of using similar carborane-based compounds for the long term prevention of estrogen dependent malignancy without inducing negative anti-estrogenic responses such as bone loss [28].
Folate metabolism is involved in processes critical in cell proliferation and DNA synthesis. The enzyme dihydrofolate reductase (DHFR) regulates cellular levels of tetrahydrofolate [3]. The dysregulation of the folate metabolic process reduces the metabolism of purine and pyrimidine, as well as the methylation of DNA, each of which are crucial processes in the cells [33]. Thus, the inhibition of the dihydrofolate reductase enzyme is an area of much interest for cancer therapy [34].
Known inhibitors of this enzyme include the anticancer agent methotrexate (MXT) 13 and the antimicrobial agent trimethoprim (TMP) 14 (Figure 3) [33,35]. Also depicted in Figure 3 is the carborane containing inhibitor 15 [35]. The DHFR inhibition activity of these compounds was measured on various cancer cell lines and their half maximal inhibitory concentrations (IC50) were reported by Reynolds and colleagues [35]. The closo-carborane derivative 15 demonstrated up to 10-fold and 100-fold greater activity in comparison with MXT 13 and TMP 14 respectively, for several of the cancer cell lines [35].
 Figure 3: MXT (13), TMP (14), and carborane DHFR inhibitor (15) [33,35]. View Figure 3
Figure 3: MXT (13), TMP (14), and carborane DHFR inhibitor (15) [33,35]. View Figure 3
The translocator protein, (TSPO) also referred to as the peripheral benzodiazepine receptor regulates the transmembrane functions of the mitochondria. The translocator protein has been shown to be overexpressed in tumor cells, making this an intriguing target for selective therapeutic and diagnostic applications [36]. Normally, TSPO actively opens and closes the pore in the mitochondria membrane. However, if the pore remains open for prolonged periods of time apoptotic factors can escape the mitochondria and enter the cytosol. This can lead to swelling of the mitochondrial matrix, interference with both ATP synthesis and oxidative phosphorylation, leading to apoptosis and necrosis of the cell [36,37].
Although this protein is a potential target for therapeutic agents, a majority of the work described in the literature focuses on targeting applications of the TPSO. Derivatives of the TPSO binding ligands have been synthesized and their IC50 concentrations were reported by Cappelli and coworkers [38]. The molecules depicted in Figure 4 represent PK11195 TPSO receptor ligand 16, isoquinolinecarboxamide derivative 17, and the carborane containing derivative 18. The binding constants and IC50 values for these ligands were 2.5 nM, 0.11 nM, and 73 nM for 16, 17, and 18 respectively. Although the TPSO binding strength and IC50 values were lower for the carborane derivative of 18, the carborane cage has the potential for incorporation of additional diagnostic and therapeutic moieties [38].
 Figure 4: PK11195 TPSO receptor ligand (16), isoquinolinecarboxamide derivative (17), and carborane derivative (18) [38]. View Figure 4
Figure 4: PK11195 TPSO receptor ligand (16), isoquinolinecarboxamide derivative (17), and carborane derivative (18) [38]. View Figure 4
Protein kinase C (PKC) is a signal transduction protein involved in tumor promotion activity when bound to phorbol esters; potent modulators of PKC are thus appealing anticancer targets [2,3,39]. PKC has been shown to modulate proliferation of cancer cells, either increasing or decreasing, dependent on the type of cancer [39]. Many studies have shown that PKC acts as a promoter of cancer cell proliferation and a suppressor of apoptosis [39]. PKC inhibitors such as benzolactam-V8-310 (BL-V8-310) 19, depicted in Figure 5, have been studied both in vivo and in vitro and their effects on various cancer cell lines have been reported [40]. In many types of cancers, inhibition of PKC resulted in a decrease of tumor growth. It is believed that this is perhaps the result of many processes including, reduced angiogenesis, increases in apoptosis, and decreases in cell proliferation [39,40]. The comparative dose response and PKC binding constants for 19 and its carborane derivative 20a-c were analyzed using bioassays of HL-60 cells. The median effective dose (ED50) values for 19 and 20 were 5 × 10-9 M and 7 × 10-9 M respectively. The binding constants for 19 and 20a-c reported as Ki = 1.8 nM and Ki = 2.0 nM, 1.4 nM, and 1.8 nM [40]. The ED50 concentrations and binding constants illustrate the potency of these carborane derivatives as PKC inhibitors, further demonstrating the potential use of carboranes in the medicinal chemistry of new molecules for the treatment of cancer.
 Figure 5: PKC inhibitor BL-V8-310 (19) and carborane derivative (20a-c) [40]. View Figure 5
Figure 5: PKC inhibitor BL-V8-310 (19) and carborane derivative (20a-c) [40]. View Figure 5
A membrane bound glycoprotein known as prostate specific membrane antigen (PSMA) is overexpressed in prostate cancer as well as newly formed vascular networks of most tumors. This cell surface protein is a target for imaging and therapy because it is expressed at levels 1,000-fold greater than in other tissues [41]. Through immune-fluorescence analysis, it has been shown biochemically that PSMA or a PSMA-antibody complex internalizes by receptor mediated endocytosis through clathrin coated pits [41].
Much work has been done in developing potent ligands for PSMA through the use of carboranes; El-Zaria and colleagues compared closo-,nido-, and iodo-carborane conjugates in vitro against known PSMA inhibitor 2-(phosphonomethyl)-pentanedioic acid (PMPA) [42]. The water soluble iodinated carborane cluster 21 (Figure 6) exhibited the highest affinity with an IC50 concentration of 73.2 nM which was comparable to PMPA with a value of 63.9 nM. However, in vivo studies exhibited lower than desired selectivity due to off target binding to areas such as the liver and gall bladder [42].
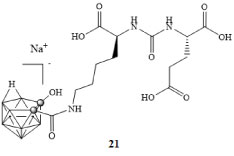 Figure 6: Water soluble iodinated carborane cluster [42]. View Figure 6
Figure 6: Water soluble iodinated carborane cluster [42]. View Figure 6
The initial diagnosis of prostate cancer is dependent on androgen levels through the androgen receptor (AR). Androgen induced transcriptional activity involves binding of testosterone 22 and 5α-dihydrotestosterone (DHT) 23 to AR. The interaction of AR with coregulators and phosphorylation of AR results in the modulation of AR transcriptional activity. Inhibition of AR and the resultant reduction of serum androgens is the basis of endocrine therapy for the treatment of prostate cancer. This has been achieved clinically using the antagonists flutamide 24 and (R)-bicalutamide 26 [43]. However, androgen ablation therapy ultimately fails and prostate cancer progresses to a hormone refractory state capable of transcriptional activity. Clinical evidence suggests the advancement of prostate cancer through several processes. These include mutations of AR that enable it to become transcriptionally active in response to ligands, alterations in expression of AR coregulators, and alterations of normal androgen axis by dysregulation of AR activity. Therefore, inhibition of AR activity and reduction of DHT concentrations (androgen ablation) may delay cancer progression [44-46].
Potential drugs that could be used to treat AR disorders include novel biologically active nonsteroidal AR ligands containing closo-carboranes [3,32]. Goto and coworkers investigated para-carborane containing ligands in developing potent AR antagonists as depicted in Figure 7 [47]. Compounds 27 and 28 exhibited lower IC50 concentrations of 3.8 × 10-7 M and 4.2 × 10-7 M, respectively, against LNCaP cells in comparison to the clinically used AR full antagonist (R)-bicalutamide 26 with an IC50 of 8.7 × 10-7 M [47].
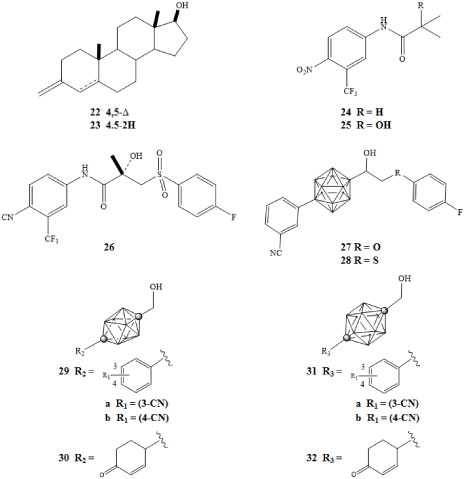 Figure 7: Testosterone (22), DHT (23), flutamide (24), hydroxyflutamide (25),
Figure 7: Testosterone (22), DHT (23), flutamide (24), hydroxyflutamide (25),
(R)-bicalutamide (26), pure AR antagonists with para-carborane cage as hydrophobic pharmacophore (27-28), ten-vertex carborane derivatives (29-30), and twelve-vertex carborane derivatives (31-32) [43,47]. View Figure 7
Fujii and colleagues developed compounds demonstrating anti-androgenic activity through novel derivatives of ten-vertex and twelve-vertex para-carborane [43]. These were tested against androgen-dependent SC-3 cells. One of the most potent ten-vertex derivatives 29a (Figure 7) bearing a 3-cyano group exhibited a higher potency than hydroxyflutamide 25 with an IC50 of 7.6 × 10-7 M [43]. Derivative 29b bearing a 4-cyano group exhibited lower affinity with an IC50 of 1.6 × 10-5 M. With regard to cyclohexanone derivatives, 30 exhibited an IC50 concentration of 6.1 × 10-6 M. The twelve-vertex derivatives 31a, 31b, and 32 exhibited IC50 concentrations of 1.9 × 10-6 M, 1.0 × 10-5 M, and 9.7 × 10-6 M, respectively. The study was a further demonstration of the potential use of carboranes as a hydrophobic core structure of biologically active molecules [43].
Purinergic receptors are signaling receptors involved in the process of neurotransmission. Activation of a specific subtype of the purinergic receptor results in the influx of Na+ and Ca2+ across the cell membrane and the release of K+ ions. The P2Y and P2X receptor subtypes have been linked to processes such as cell proliferation, differentiation, and apoptosis [48]. When exposed to high concentrations of ATP for a sufficient period of time, the cation channel of the P2X7 receptor can be converted to a nonspecific transmembrane pore that allows the passage of small molecules as large as 900 daltons [49]. This process along with the involvement of the caspase enzyme system has been associated with apoptosis of the cell [48,50].
The P2X7 receptor has been implicated in ATP mediated cell death, regulation of receptor trafficking, and inflammation; it is a potential target for cancer therapies. An agonistic response of the receptor is required to facilitate apoptosis. BzATP is one such molecule that has a larger binding constant than that of ATP to the binding site of the P2X7 receptor and has been used in assays determining the state of the transmembrane pore associated with P2X7 activation [48,51]. Adenosine 33 and 2'-deoxyadenosine 34 are known agonists of the P2X7 receptor. Olejniczak and colleagues prepared the carborane derivatives 35-37 shown in Figure 8 [52]. The carborane derivatives 35-47 each had a greater agonistic effect than 33 or 34 [53].
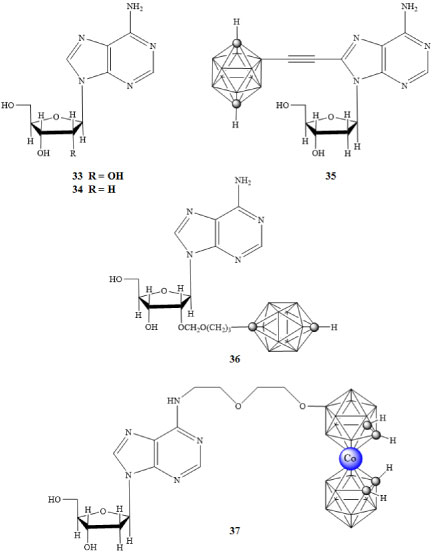 Figure 8: Purinergic receptor agonists adenosine (33) and 2'-deoxyadenosine (34), and carborane containing P2X7 agonist derivatives (35-37) [52,53]. View Figure 8
Figure 8: Purinergic receptor agonists adenosine (33) and 2'-deoxyadenosine (34), and carborane containing P2X7 agonist derivatives (35-37) [52,53]. View Figure 8
NAD+ is a vital cofactor used throughout cellular respiration. In addition to its role in glycolysis, the TCA cycle, and the reaction linking this cycle with oxidative phosphorylation, NAD+ is consumed as a substrate by more than 300 known NAD-dependent enzymes. Mammals possess a unique NAD+ recycling pathway, which converts nicotinamide to NAD+. The first and rate-limiting step in this pathway is catalyzed by the enzyme Nicotinamide phosphoribosyltransferase (Nampt). Nampt has been shown to be upregulated in many/most cancers [54-61] and its overexpression is highest in aggressive and refractory cancers, making it a promising target for the treatment of various cancers [62,63].
FK866 38 is the first known small molecule inhibitor of Nampt and has been investigated in several Phase I/II clinical trials against several cancers [64,65]. Recently, we reported the synthesis and anticancer activity of several carborane-containing derivatives, where the benzoylpiperazine of 38 was replaced by a number of different moieties, including adamantyl, ortho-, meta-, or para-carborane [66,67]. Of the new agents reported, a derivative containing a meta-carborane 39 exhibited a 100-fold greater inhibition of Nampt and a 10-fold increase in potency with respect to FK866 in several human cancer cell lines in vitro.
The carborane-based inhibitor 39 demonstrated low, to sub-nanomolar potencies against human lung, colon, and breast cancers in vitro and the hydrophobic carborane moiety was shown to significantly increase agent potency over other, similarly sized organic moieties [66,67]. Recently, the synthesis of a new derivative of 39, bearing a hydroxymethyl group attached to the carborane cluster was reported, allowing for the covalent attachment of the drug to various targeting groups through the use of facile click chemistry reactions [66,67] (Figure 9).
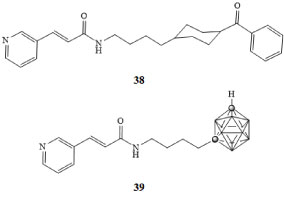 Figure 9: FK866 (38) and meta-carborane derivative of FK866 (39) [66,67]. View Figure 9
Figure 9: FK866 (38) and meta-carborane derivative of FK866 (39) [66,67]. View Figure 9
The relationship between hypoxia inducible factors HIF-1α and HIF-2α has been an area of much interest. The levels of these proteins are closely related to the oxygen content of the cells [68]. These proteins are expressed in low oxygen environments and have been linked to angiogenesis [69]. Due to the higher metabolic rates of cancer cells, tumors typically have a lower oxygen content than normal tissues, resulting in increased levels of HIF-1α and HIF-2α in cancerous tissues [68]. Others have probed the relationship between tumor growth and metastasis and many of these studies suggest that these proteins have a profound effect on the survival and growth of cancerous tissues [69]. This protein has therefore been a target of interest for the treatment of various types of cancer. Nakamura and colleagues developed carboranyl derivatives of the LW6 drug molecule 40 depicted in Figure 10 for HIF-1 inhibition [70]. Activity of these molecules was measured in HeLa cells and growth inhibition IC50 concentrations were reported [70]. The carborane derivative GN26361 41 exhibited a 10-fold increase in activity compared with LW6 40. The results of this study support the use of carborane derivatives for the design of inhibitors of this protein. In principle, molecules such as these could be used to combat the angiogenesis of growing tumors [71,72].
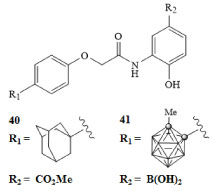 Figure 10: Hypoxia inducible factor HIF-1α inhibitor LW6 (40) and carborane derivative GN26361 (41) [70]. View Figure 10
Figure 10: Hypoxia inducible factor HIF-1α inhibitor LW6 (40) and carborane derivative GN26361 (41) [70]. View Figure 10
The Tumor Necrosis Factor α (TNF-α) is a protein involved in the early steps of a complex pathway that has been linked to inflammatory responses and cell survival including apoptosis. There are multiple ways in which the TNF-α protein can promote tumor growth. The TNF-α interacts with adjacent cells in an autocrine manner. Autocrine processes have been linked to increased damage of genetic material in malignant cells or cells with malignant potential, as well as the improved survivability of malignant cells. The expression of TNF-α leads to the production of cytokines and chemokines which when combined, can lead to an angiogenic response [2,73]. The inhibition of this protein and the resulting effects on cancer cell growth have been explored [74,75]. Phthalimides are regulators of the TNF-α protein, under specific conditions these molecules have been shown to inhibit the production of TNF-α and therefore have been explored to treat a variety of diseases including cancers [73]. The N-phenylphthalimides 42a-c and adamantyl substituted phthalimide 43 drug molecules along with carborane derivatives of these phthalimide compounds, depicted in Figure 11, were prepared and TNF-α inhibition was measured [76]. The N-(1,2-dicarba-closo-dodecaboran-1-yl-methyl) phthalimide 44 and the N-(1,2-dicarba-closo-dodecaboran-1-yl)-4,5,6,7-tetra-fluorophthalimide 45 were analyzed to determine their relative potency for TNF-α inhibition [76]. The carborane derivative 45 demonstrated a 100-fold greater inhibitory response when compared with 42a-c and 43 [76].
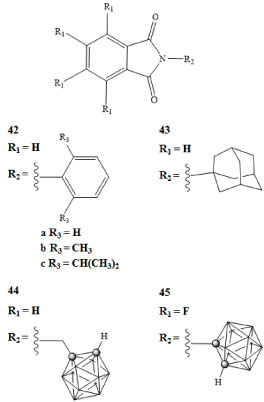 Figure 11: Phthalimide compounds used in TNF-α regulation (42-43) and carborane derivatives of phthalimides (44-45) [76]. View Figure 11
Figure 11: Phthalimide compounds used in TNF-α regulation (42-43) and carborane derivatives of phthalimides (44-45) [76]. View Figure 11
The retinoic acid receptor (RAR) and retinoid X receptor (RXR) have recently been implicated in the regulation of gene networks involved in the control of cell growth, differentiation, survival, and death. These two receptor proteins, when bound to their corresponding ligands, form the RAR-RXR heterodimer [3,77]. The RAR and RXR binding ligands include all-trans-retinoic acid 46 and 9-cis-retinoic acid 49 respectively. The RAR-RXR heterodimer can be activated by an RAR agonist, such as AM80 47, but not by an RXR agonist. The presence of an RXR agonist, such as PA024 48, can increase activity of an RAR agonist and thus promote transcriptional activity of the RAR-RXR heterodimer, even when RAR agonist concentrations are low [77,78]. The retinoid X receptor can form heterodimers with other nuclear receptors; some of these heterodimers can become transcriptionally active in the presence of RXR ligands alone [79].
The amount of cell differentiation and the half maximal effective concentrations (EC50) of the carborane containing agonist derivatives 51a-f were reported [78]. The EC50 for the carborane derivative 51d shown in Figure 12 was 1.5 × 10-9 M, which is comparable to the activity of the all-trans-retinoic acid [78]. The activities of carborane RXR antagonists 52a-e were measured in HL-60 cancer cells in the presence of the RXR and RAR binding ligands Am80 and PA024, both at concentrations of 1 × 10-10 M. The inhibition of cell differentiation was measured and EC50 concentrations were reported. The carborane derivative 52b exhibited higher potency than the RXR antagonist PA452 50 by almost an order of magnitude [78].
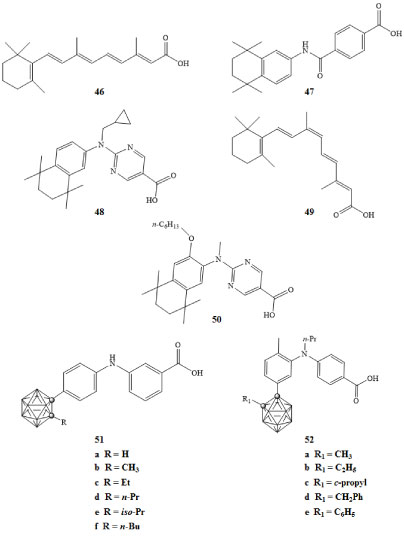 Figure 12: RAR binding ligand all-trans-retinoic acid (46), RAR agonist Am80 (47), RXR agonist PA024 (48), RXR binding ligand 9-cis-retinoic acid (49), RXR antagonists PA452 (50), carborane derivatives of RAR agonists (51a-f), and carborane derivatives of RXR antagonists (52a-e) [78]. View Figure 12
Figure 12: RAR binding ligand all-trans-retinoic acid (46), RAR agonist Am80 (47), RXR agonist PA024 (48), RXR binding ligand 9-cis-retinoic acid (49), RXR antagonists PA452 (50), carborane derivatives of RAR agonists (51a-f), and carborane derivatives of RXR antagonists (52a-e) [78]. View Figure 12
Recent work has clearly demonstrated the immense potential that carboranes have as pharmacophores in the development of new, small molecules for the treatment of various types of cancers. The synthetic flexibility of carboranes, along with their rigid structure and chemical stability, make them appealing moieties for use in cancer drug discovery.