S100A8 and S100A9 Ca2+ binding proteins influence a wide range of cellular processes, including cell differentiation, tumorigenesis, and inflammatory and autoimmune disorders. Both proteins are constitutively expressed in early myeloid lineage, with reductions during myeloid cell differentiation and maturation. Under normal conditions, S100A8 and S100A9 are present in circulating monocytes and granulocytes, but not resting tissue macrophages. During the stress-mediated response to infection and tissue injury, their levels markedly increase and contribute to acute and chronic inflammatory disorders as cell autonomous and non-autonomous activators of toll-like receptors (TLRs). There is controversy, however, whether S100A8 and S100A9 are pathogenic or protective during infection and inflammation. Some studies suggest that S100A8 and S100A9 proteins function extracellularly to amplify TLR-mediated responses, thereby increasing inflammation and tissue damage. Others support an adaptive anti-inflammatory role during acute infection and inflammation. This review focuses on the immunobiology of S100A8 and S100A9 in acute systemic inflammation induced by sepsis.
The S100A8 and S100A9, also known as myeloid-related protein 8 (MRP8) and MRP14, are members of the S100 protein family [1-3]. Like other S100 proteins, which includes 25 family members, S100A8 and S100A9 have two conserved Ca2+ binding domains connected by a variable hinge region thought to regulate its biological activity and is distinct for each family member [4,5]. S100A8 and S100A9 are constitutively expressed in early myeloid lineage, with reductions during myeloid cell differentiation and maturation, but low protein levels are maintained in circulating monocytes and neutrophils but not resting tissue macrophages [1]. Both S100A8 and S100A9 are also expressed in other cell types after activation, including platelets, esteoclasts, keratinocytes, and vascular endothelial cells [1]. Under cellular stress conditions, such as infection and inflammation, S100A8 and S100A9 proteins are induced by a variety of mediators and form homodimers and heterodimers in the cytosol in a Ca2+- dependent manner [2,3]. Because of their Ca2+ binding properties and high expression levels in activated granulocytes, S100A8 and S100A9 are also termed Calgranulins [6]. Their heterodimer complex is called Calprotectin because of its protective, anti-microbial effects [7].
Early studies show that S100A8 and S100A9 can be proinflammatory mediators, causing acute inflammation [3,8,9] by binding to and activating the toll-like receptor 4 (TLR4) and by amplifying the stimulatory effect of bacterial products, e.g., endotoxin/ LPS, thus causing excessive inflammation and tissue damage. Subsequent studies support that S100A8 and S100A9 can also play an anti-inflammatory role during infection and inflammation [1]. This emerging adaptation is supported by the finding that S100A8 and S100A9 expression can be induced by anti-inflammatory mediators such as IL-10 [10], and that they can exert direct anti-microbial activity [2]. These multifunctional roles of S100A8 and S100A9 may reflect differences in their intracellular and extracellular activities, depending on the inflammatory milieu [1,2]. This review will discuss both the proinflammatory and anti-inflammatory roles of S100A8 and S100A9 proteins, with an emphasis on sepsis-driven inflammation.
Like most S100 proteins, S100A8 and S100A9 are located on chromosome 3 in mice and chromosome 1q21 in humans [11,12]. Structurally, S100A8 and S100A9 proteins have two helix-loophelix, Ca2+ -binding domains connected by a hinge region [3]. The C-terminal domain has a higher affinity to Ca2+ binding [3,11,13]. S100A8 and S100A9 are either constitutively expressed or inducible depending on the cell type and the environmental stimuli [1,3]. S100A8 and S100A9 are constitutively expressed in myeloid lineage cells and expression correlates with the state of differentiation [14,15]; expression decreases as immature myeloid cells differentiate into monocytes and dendritic cells [16,17]. In distinct contrast, mature human neutrophils constitutively express high levels of both proteins [18]. Circulating monocytes constitutively express low levels of S100A8 and S100A9 proteins which diminish upon monocyte extravasation from blood and subsequent differentiation into macrophages in tissues, while normal/resting macrophages do not express both proteins [17,19].
Both S100A8 and S100A9 expressions increase in response to bacterial products such as LPS, proinflammatory cytokines such as TNFα and IL-1β, and anti-inflammatory cytokines such as IL-10 and TGFβ [1,20,21]. These seemingly disparate responses occur in acute and chronic inflammatory conditions with activated neutrophils and macrophages expressing high levels of S100A8 and S100A9 [1,22,23]. The immunosuppressive cytokine IL-10 induces S100A8 and S100A9 expression in differentiated human dendritic cells [24] and in endotoxin-tolerant monocytes [10], which in turn synergize with bacterial LPS to promote S100A8 expression in murine macrophages [25]. The CCAAT/enhancer-binding protein C/EBPβ has been shown to regulate S100A9 transcription in myeloid cells upon LPS stimulation [26]. This transcription factor has also been implicated in the induction of the S100A8 promoter in keratinocytes in response to IFNγ stimulation [27]. Stat3 also supports transcription of S100A8 and S100A9 in myeloid cells [28], and several Stat3 binding sites locate on the S100A8 and S100A9 promoters [16].
Multiple processes also control S100A8 and S100A9 protein secretion/release. Both form non-covalent homodimers and heterodimers dependent on Ca2+ binding and protein kinase C (PKC) activation [29,30], with Ca2+ binding increasing heterodimer stability [31]. At high calcium levels, the heterodimer formation is the dominant form and is more stable than the homodimers [3]. In mice, S100A8 and S100A9 exist as homodimers and heterodimers [3,32] and in humans as monomers, heterodimers and heterotetramers [3,33]. However, mouse and human S100A8 and S100A9 are considered functionally homogeneous, i.e., having similar functions [34,35]. Homodimer and heterodimer formation can affect some functions of S100A8 and S100A9 such as chemotactic effects and cytoskeleton organization, suggesting that protein dimerization mediates some of the biological effects of S100A8 and S100A9 [1]. S100A8/S100A9 heterodimerization may increase interactions with target proteins since it generates one binding site on each side of the two monomers in the heterodimer complex [36]. An increase in Ca2+ levels promotes translocation of S100A9 from the cytosol to the plasma membrane by a protein kinase C-dependent mechanism [37]. S100A8 and S100A9 lack a secretion signal, and controversy exists regarding their mode of secretion. Recent data demonstrate that both S100A8 and S100A9 are not secreted via the classic Golgi-dependent pathway but rather by an energy-dependent pathway that requires an intact cytoskeleton [38], activation of PKC, and formation of microtubules but not de novo protein synthesis [3,38]. This mode of secretion is mainly observed in activated monocytes/macrophages and neutrophils and accounts for most of the extracellular S100A8 and S100A9 proteins at inflammation sites [8,39]. Also, passive release at inflammation sites of S100A8 and S100A9 from activated neutrophils dying from necrosis may occur [39]. S100A8 and S100A9 can also be released from activated neutrophils during neutrophil extracellular trap formation, which facilitates antimicrobial activity [40]. Furthermore, post-translational modification of S100A9 via phosphorylation at Threonine 113 by p38 MAPK has been observed in activated human neutrophils [41]. Although this phosphorylation does not affect the heterodimer formation, it may regulate interaction of the S100A8/ S100A9 heterodimer with the microtubules, which is critical for neutrophil migration [9]. Because secretion of the S100A8/S100A9 heterodimer also depends on functional microtubule polymerization [8,38], it remains to be seen whether S100A9 phosphorylation by p38 MAPK supports heterodimer secretion. In this context, a recent study showed that stimulation of human monocytes with IL-10 increased S100A8/S100A9 heterodimer more efficiently than TNFα, but enhanced secretion of the heterodimer required both TNFα and IL- 10 signals [42]. This report suggested that IL-10 signal may activate p38 MAPK to phosphorylate S100A9 in order to efficiently secrete S100A8/S100A9 heterodimer.
The murine S100A9 protein constitutes 10-20% of neutrophil cytosolic proteins, whereas the human S100A8/S100A9 heterodimer complex constitutes 45% of cytosolic proteins in neutrophils and ~1% in monocytes [33]. When released in the extracellular space, S100A8 and S100A9 are chemotactic for murine and human macrophages and neutrophils [32,43,44], which is required for microbial clearance [2]. Neutrophils, followed by macrophages, are rapidly recruited to sites of acute inflammation by bacterial chemoattractants and activated complement components such as C5a [2]. Release of S100A8 and S100A9 proteins by activated phagocytes then recruit more phagocytes to the infection site. Lackmann and colleagues [32] first reported that murine S100A8 is chemotactic for neutrophils and monocytes in vivo by showing that injection of S100A8 into the mouse food pad promotes phagocyte recruitment, with early recruitment of neutrophils at 4-6 hours followed by monocytes over 24 hours.
Mechanistically, phagocyte recruitment to infection and inflammation sites requires that leukocytes transmigrate to activated endothelium, which is facilitated by binding of selectins on endothelial cells to glycoproteins on leukocytes [45]. Subsequent interaction of leukocyte integrins with the vascular endothelial cell adhesion molecules facilitates adherence and immobilization of leukocytes [46,47]. Some studies suggest that S100A8/S100A9 heterodimers released from transmigrating neutrophils at local infection sites can amplify leukocyte recruitment, via facilitating leukocyte-endothelial cell interaction [48]. In support of this, human S100A8 and S100A9 increase β2 integrin CD11b expression and affinity on phagocytes [49]. In this scenario, S100A9 heterodimerizes with S100A8 as reported using recombinant S100A9 to study leukocyte-endothelial adherence interactions by neutrophils [43] and monocytes [50]. Moreover, murine S100A9 null neutrophils migrate in vitro in response to the chemokine IL-8 despite failing to upregulate CD11b expression [51]. However, leukocyte recruitment in response to thioglycollate-induced peritonitis is normal in S100A9 null mice [52], and these mice are resistant to LPS-induced endotoxemia [9]. In contrast, antibody blockade of S100A8 and S100A9 in a mouse model of Streptococcal pneumoniae strongly inhibits neutrophil and macrophage recruitment to the alveoli but has no effect on clearing bacterial load [53]. This defect may be due to S100A8 ability to regulate the microtubule network in response to Ca2+ elevation, a step required for leukocyte migration [3,54]. In contradiction, a recent study showed that leukocyte recruitment was not affected in S100A9 null mice; S100A8 degrades in the absence of its binding partner, S100A9 [52]. As another biological property, S100A9, or S100A8/ A9 heterodimers may regulate cell signaling and affect leukocyte migration. For example, human monocyte infiltration is reduced when S100A9 phosphorylation at threonine 113 is blocked by the p38 MAPK inhibitor, suggesting that this phosphorylation may affect its heterodimer complex formation with S100A8 [54,55]. Table 1 lists the main biological functions described for S100A8 and S100A9 so far and their association with inflammatory diseases/conditions. It is evident that S100A8 and S100A9 broadly participate in inflammation, whether acute or chronic, proinflammatory or anti-inflammatory.
Table 1: S100A8/S100A9 functions and association with inflammatory conditions. View Table 1
S100A8 and S100A9 proteins are damage-associated molecules that initiate inflammatory responses by binding to and activating damage-associated molecular pattern (DAMP) sensors receptors such as TLR4 and the receptor for advanced glycation end products (RAGE) on innate immune cells [3,81]. Extensive work by Roth and colleagues [3,9] indicate that S100A8 and S100A9 act as endogenous danger signals to amplify inflammatory responses to infection and injury. S100A8/S100A9 heterodimer promotes endotoxin-induced shock and lethality in mice [9], and mice lacking S100A8 and S100A9 expression resist LPS endotoxemic reactions and E. coliinduced abdominal sepsis [9]. Furthermore, bone marrow-derived myeloid cells lacking S100A8 and S100A9 proteins exhibited reduced responses to LPS, as demonstrated by reduced production of the proinflammatory mediators TNFα, IL-6, and IL-8. This defect was restored by the stimulation with recombinant S100A8 and S100A9 proteins, thus suggesting that they can function as proinflammatory mediators [9].
When stimulated, TLR4 on innate immune cells triggers signaling cascades that include MAP kinases and NF-κB transcription factors that increase proinflammatory cytokines and chemokines [82-84]. S100A8/S100A9 protein complexes act as endogenous activators of TLR4 to promote immune responses to infection [9,85]. Strong evidence for this path is the work of Vogl T [9] and van Zoelen [67], which showed that S100A8 homodimers and S100A8/S100A9 heterodimers directly activate TLR4 on murine macrophages, and that S100A8 protein is the active component that binds specifically to the TLR4-MD2 receptor complex. This molecular bridge couples with adapter protein MyD88 and sequential activation of IRAK1, ERK, p38 MAPK and NF-κB, and production of proinflammatory cytokines and chemokines [9,67]. Since infections and bacterial toxins such as LPS induce S100A8 and S100A9 protein expression and release [8,9,67], S100A8 and S100A9 can amplify the ongoing innate immune and inflammatory responses [3,86]. In support of this, S100A9 null mice, which also lack S100A8 protein but not mRNA due to enhanced S100A8 protein metabolism, exhibit reduced acute/systemic inflammatory response to infection as demonstrated by the reduced production of proinflammatory cytokines such as TNFα [8,67]. The findings that LPS stimulation in murine phagocytes promotes S100A8 and S100A9 production while S100A9 null phagocytes exhibit attenuated response to LPS support that S100A8/ S100A9 protein complexes function in paracrine and autocrine pathways to amplify acute inflammatory responses [8,9,67]. Indeed, TLR-4 mediated stimulation by LPS and S100A8 activate the same signaling routes since murine phagocytes lacking functional TLR4 receptors did not respond to the stimulation with the S100A8/ S100A9 complex [3,8]. In addition, S100A8 and S100A9 expression reflects various inflammatory autoimmune disorders [87,88]. In a mouse autoimmune disease model, local release of S100A8 and S100A9 induced autoreactive CD8+ T cells and enhanced a systemic autoimmune reaction mediated via TLR4 [85].
In addition to TLR4, the RAGE receptor, which binds glycation end products (N-glycans) and other unrelated ligands, is a target receptor for S100A8 and S100A9 [89,90], and recent reports implicate RAGE in the acute inflammatory responses [91]. Similar to TLR4, the binding of S100A8 and S100A9 to RAGE activates the signaling cascades that lead to MAPK and NF-κB activation [89]. This binding depends on RAGE carboxylated glycans [92]. In this context, S100A9 appears responsible for the S100A8/S100A9 heterodimer binding to RAGE because the S100A9 homodimer shows greater binding affinity than the S100A8/S100A9 heterodimer in phagocytes stimulated in vitro [93]. This property contrasts with TLR4 activation by the S100A8/S100A9 heterodimer where the S100A8 component promotes the S100A8/S100A9 heterodimer binding to the TLR-MD2 receptor complex [9]. Because TLR4 and RAGE are co-expressed on many innate immune cells, including phagocytes, it is unclear which receptor is more important in the inflammatory response to S100A8/ S100A9. It has been argued that one receptor at a time likely mediates S100A8/S100A9 signaling and that the differential effect and receptor engagement may depend on the cell type, pathophysiological context, and the oligomerization state of S100A8 and S100A9 proteins [2,3]. Given that S100A8 and S100A9 activate phagocytes at very low molar concentration, it is possible that they synergize to amplify the inflammatory response under certain conditions by activating both TLR4 and RAGE. Thus, S100A8 and S100A9 are proinflammatory mediators that may promote defense at adjacent or distant tissue injury. Figure 1 shows a suggested scheme of pathways that induce S100A8/S100A9 expression and secretions, as well as their intracellular and extracellular effects on innate immunity cells.
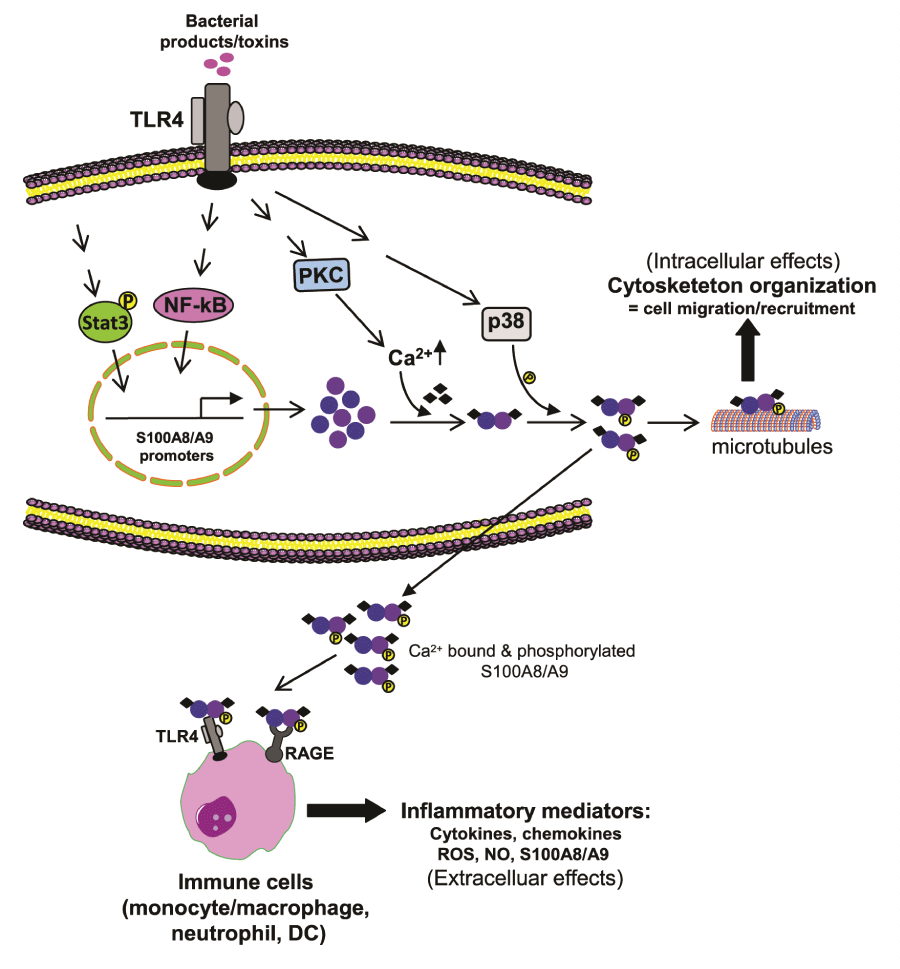 Figure 1: Expression and activity of S100A8/S100A9.
Figure 1: Expression and activity of S100A8/S100A9.
During infection, bacterial toxins activate TLR4 to induce S100A8/S100A9 expression in phagocytes. Protein kinase C increases Ca2+ levels, which dacilitates
S100A8/S100A9 protein heterodimerization. Activation of p38 MAPK phosphorylates S100A9 and also facilitates heterodimerization as well as secretion.
Intracellularly, the S100A8/S100A9 binds to the actin filaments, which promotes tubulin polymerization and bundling of microtubules, thus leading to cytoskeleton
reorganization phagocyte migration, and recruitment. When released extracellularly, S100A8/S100A9 binds to and activates TLR4 and RAGE receptors on
phagocytes and other innate cells. These cells then release inflammatory mediators to amplify the ongoing inflammatory responses. S100A8/S100A9 also
activates its own expression, thus acting in a paracrine and autocrine manner.
View Figure 1
Although most studies initially demonstrated proinflammatory effects of S100A8 and S100A9, recent reports suggest that S100A8 and S100A9 also can play anti-inflammatory roles during infection and inflammation. Ikemoto et al. [65] found that the S100A8/S100A9 heterodimer exerts an anti-inflammatory effect by reducing acute inflammation and liver injury during LPS-induced endotoxemia; endotoxemic rats intraperitoneally-injected with S100A8/S100A9 protein complexes purified from human neutrophils produced smaller amounts of the proinflammatory mediators IL-6 and nitric oxide (NO) and had less liver damage compared with rats injected with saline. Another study demonstrated that intraperitoneal delivery of recombinant S100A8 and S100A9 in endotoxemic mice reduced neutrophilic infiltration and attenuated liver, kidney, and lung injury [69]. These studies suggest that S100A8 and S100A9 in certain contexts can attenuate acute proinflammatory responses to bacterial toxins. Using a rat model of autoimmune myocarditis, Otsuka et al. [70] showed that intraperitoneal injection of recombinant S100A8/S100A9 reduced myocardial inflammation and damage by attenuating the expression of the proinflammatory cytokines TNFα, IL-1β, and IL-6.
Extracellular S100A8 and S100A9 complexes also may exert direct antimicrobial activity to confer resistance to bacterial invasion. Surprisingly, purified S100A8 and S100A9 have broad antibacterial activity in vitro against microbes, including Candida albicans, E. coli, and Staphylococcus aureus [2]. This effect depends on chelating the divalent cation Zn2+. The Zn2+-binding sites in S100A8 and S100A9 are structurally conserved and, like Ca2+-binding sites, induce heterotetramer complex formation [31]. Zn2+ is an essential nutrient for bacterial growth [94], and by the binding to and sequestration of the Zn2+ present at local inflammatory sites, S100A8/S100A9 complexes can inhibit bacterial growth [95]. In support of this observation, moderate increases in Staphylococcus aureus infection occur in S100A9 null mice, which coincide with higher levels of Zn2+ within the abscesses [96]. Human S100A8 and S100A9 normally occur in airway secretions [97] and in inflammed gastric mucosa [98], suggesting that they may function to limit growth of commensal organisms and defend against pathogen invasion. In contrast, a recent study reported that S100A8/S100A9 complex had no antimicrobial effects during E. coli-induced urinary tract infections, as bacterial growth, neutrophil infiltration, and inflammatory cytokine production were similar in wild type and S100A9 null mice [99]. Furthermore, during infection, phagocytes produce high levels of intracellular anti-infective reactive oxygen species (ROS) and anti-inflammatory reactive nitrogen species (RNS) for microbial killing [5], although ROS production at inflammation can cause tissue damage. In this light, S100A8 and S100A9 may function as ROS scavengers to reduce inflammationdependent oxidative damage [100]. S100A8 and S100A9 may also play an anti-inflammatory role via nitric oxide (NO) production. They can induce NO synthase to increase NO levels in murine macrophages [101]. NO promotes vascular homeostasis and anti-microbial defense [102], and S100A8 can act as an NO shuttle via covalent binding [62].
While most studies described anti-inflammatory roles for extracellular S100A8 and S100A9, recent studies by Gabrilovich and colleagues [16] and Chen et al. [103] suggest that intracellular S100A8 and S100A9 can have profound effects on immune and inflammatory responses via targeting hematopoiesis and myeloid cell differentiation. They showed that Stat3-inducible upregulation of S100A9 enhances MDSC generation in tumor-bearing mice. MDSCs are precursors of monocytes, granulocytes and dendritic cells and possess potent immunosuppressive activities during infection and inflammation [104-106]. S100A9 null mice, which simultaneously lack the S100A8 protein, generate fewer MDSCs and mount potent anti-tumor immune responses, whereas over expression of S100A9 in embryonic stem cells inhibits macrophage and dendritic cell differentiation leading to MDSC accumulation [16]. S100A9 binds CD33 receptors on MDSCs from patients with myelodysplasia and induces anti-inflammatory/immunosuppressive cytokines IL-10 and TGFβ production by MDSCs [103].
Acute sepsis, initiated by infection or trauma, is characterized by systemic inflammation due to activation of TLR4 and excessive production of proinflammatory cytokines and chemokines, such as TNFα, IL-1β, IL-6, and IL-8, which cause vascular dearrangement, shock, and multiorgan failure [107-109]. The initial so-called systemic circulation cytokine "storm" is almost simultaneously epigenetically and post-translationally reprogrammed to a protracted adaptation state with persistently repressed innate and acquired immune responses. Among the immune changes associated with this cellular reprogramming and immunosuppression are increases in IL-10 and TGFβ production, increases in repressor T regulatory cell numbers, and dendritic cell apoptosis. This reactive adaptor phenotype delays clearance of the primary inciting infection and increases risk for secondary bacterial and viral infection [108].
Recent studies by Vogl et al. [9] and van Zoelen et al. [67] showed a link between S100A8 and S100A9 protein expression and sepsis pathophysiology and suggested that they can play a pathogenic, proinflammatory role at least during sepsis initiation. This study found that levels of the S100A8/S100A9 protein complex were elevated systematically and locally in mice during E. coli-induced abdominal sepsis [67]. This response was associated with increased production of TNFα and IL-6 and enhanced bacterial dissemination, whereas S100A9 null mice had reduced bacterial dissemination and were protected from acute sepsis inflammation [9,67]. In addition, patients with peritonitis-derived sepsis had very high levels of S100A8/S100A9 complexes both in the circulation and the abdominal fluid at days 1-3 after sepsis onset [67]. These studies suggest that S100A8/S100A9 proteins play a proinflammatory role in sepsis. In contrast, S100A8/S100A9 complexes can become anti-inflammatory and protective during Gram-negative pneumonia-derived sepsis [68]. Infection with Klebsiella pneumoniae in S100A9 null mice, which also lacks the S100A8 protein, results in significant increases in TNFα, IL-1β, and IL-6, enhanced bacterial dissimination 48 hours after infection, increased organ damage, and reduced survival, suggesting that S100A8/S100A9 complexes exert an anti-inflammatory effect Gram-negative pneumonia-derived sepsis. In septic patients, a recent study suggested that protracted elevated S100A9 mRNA levels can predict hospital-acquired infections [79]. S100A9 mRNA levels were elevated in whole blood leukocytes from sepsis patients. Interestingly, mRNA levels decreased at days 7-10 in patients that did not progress to late sepsis, but remained elevated in patients that later acquired opportunistic infections and entered the late phase, i.e., were immunosuppressed [79]. In this study, the functional/secreted S100A9 protein level was not measured and thus the results may not indicate an actual S100A9 effect during the late, anti-inflammatory septic phase. The same group [10] also reported that S100A8 and S100A9 mRNA levels were increased in normal blood mononuclear cells that were rendered endotoxin-tolerant by exposure to a low dose of bacterial endotoxin/LPS. Of note, endotoxin tolerance observed in circulating leukocytes from animals and humans with late sepsis, is associated with unresponsiveness to bacterial toxins and suggests immunosuppression [110,111]. In addition, a recent study in septic shock patients showed that S100A8 mRNA and protein levels decreased along the recovery [78]. The S100A8 protein decreased in blood mononuclear cells 7 days after sepsis diagnosis in patients that recovered. However, the protein level decreased even further in patients that did not survive the septic shock [78]. Because only a limited number of 17 patients were used in this study and the mononuclear cell preparation also contained lymphocytes, it is unclear from these results if the decrease in S100A8 protein during the late sepsis phase can predict survival. Thus, although the above studies indicate association between S100A8 and S100A9 expression and sepsis pathogenesis, further studies are needed to clarify whether these proteins promote or attenuate inflammation during sepsis. Together, the published data support a dual proimmune and antiimmune function of S100A8 and S100A9 in sepsis.
Within the immune system, S100A8 and S100A9 are expressed under normal conditions in neutrophils and at very low levels in macrophages, suggesting that they may play a role in immune homeostasis, e.g, via maintenance and organization of the cytoskeleton. However, the findings that expression of S100A8 and S100A9 is dramatically increased during acute and chronic inflammation indicate that they play multifunctional roles during cellular stress-induced responses to infection and inflammation. While support for their proinflammatory functions as amplifiers of the immune and inflammatory responses during acute inflammation mounting, emerging data suggests they can be anti-inflammatory and directly antimicrobial protective mediators. Specific changes in the inflammatory microenvironment most likely play an important role in the functional diversity of S100A8 and S100A9. The posttranslational modifications on S100A8 and S100A9 proteins, transition metal (Zn2+,Ca2+) binding, protein complex formation, and cell autonomous and non-autonomous activities indicate their pleiotrophic function in inflammation and immunity. In addition, receptor binding and co-stimuli may also play a role in regulating the pro- and anti-inflammatory aspects of S100A8 and S100A9 functions. There is no distinct receptor identified for S100A8 and S100A9 thus far; both proteins bind and activate the TLR4 and RAGE receptors, which also are receptors for many other inflammatory mediators. Sepsis pathophysiology involves both proinflammatory and antiinflammatory responses, which are influenced by S100A8 and S100A9. Thus, better understanding of the mechanisms that regulate the biology and function of S100A8 and S100A9 may inform new treatment targets for inflammatory diseases.