Spinocerebellar ataxia 2 is a rare neurodegenerative disease characterized by a complex neurological presentation of ataxia of adult onset with an autosomal dominant inheritance caused by an abnormal expansion of trinucleotide CAG repeat.
The aim of this study is to determine the correlation of CAG trinucleotide repeat length with Scale for assessment and rating of ataxia score, age of onset and disease duration among genetically-positive individuals.
The study involved 10 genetically-positive spinocerebellar ataxia 2 individuals in a Filipino family. CAG trinucleotide repeat length were determined using genetic studies done in Yokohama University. A statistical analysis of the results were performed using Pearson's and Spearman's rho correlational coefficient. The statistical significance level was established for p-values < 0.05.
Among the 10 individuals, the mean age of disease onset is 46.8 ± 11.7 years, disease duration of 10.2 ± 6.3 years and SARA scores of 15.2 ± 12.5. There is a significant inverse correlation of CAG trinucleotide repeat length and age of onset (Pearson correlation coefficient of r = -0.778 (p-value = 0.008) and Spearman's rho of -0.932 (p-value < 0.001). Disease duration and SARA scores did not show any significant correlational analysis with CAG trinucleotide repeat lengths.
This is the first study on spinocerebellar ataxia 2 in a Filipino family with 10 genetically-confirmed individuals exploring the genotype and phenotype characteristics. The findings of this study confirmed the complex symptomatology of spinocerebellar ataxia 2. Further studies are recommended to further elucidate the diverse involvement of the nervous system to pave way for novel disease-treatment.
Spinocerebellar ataxia 2, CAG trinucleotide repeat length, Scale for assessment and rating of ataxia score
SCA: Spinocerebellar Ataxia; SARA: Scale for Assessment of Rating and Ataxia Score
Spinocerebellar ataxia (SCA) is a neurodegenerative disorder associated with trinucleotide repeat mutations and sharing a complex neurological presentation of an ataxia of adult onset and an autosomal dominant inheritance [1-5]. After SCA3, SCA2 is noted to be the second most prevalent type worldwide, accounting for 15% of all cases [2].
It is known that the disease with a mean progression rate is 1.49 per year at the Scale for Assessment and Rating of Ataxia (SARA) [6-9]. As with variable presentation of this disease, there is still no definite treatment for SCA2 [10,11]. To date, there is no available data regarding the prevalence or incidence of SCA2 in the Philippines nor studies involving this disease in a Filipino subset. Furthermore, there are a few published case reports of autosomal dominant SCA (SCA7, SCA13 and SCA25) afflicting Filipino families [12-15].
Furthermore, there are no correlational studies on the genotype-phenotype variability of SCA in Filipinos. This is the first study on Filipinos afflicted with SCA2 which aims to describe the clinical features of 10 genetically-tested Filipino patients, and to determine the correlation of CAG repeat lengths of each affected individual with age of onset, disease duration and SARA (Scale for the assessment and rating of ataxia) scores.
This is a retrospective study approved by the Institutional Review Board, initiated following the examination of the proband who presented with features of gait imbalance and slurring of speech, and provided a family history of the same illness and tested positive for SCA2 gene, confirmed in Yokohama University (Figure 1). A total of 24 symptomatic relatives were noted in the family tree generated through history taking. However, 9 of these symptomatic relatives were deceased. Out of the 15 living symptomatic relatives, only 10 gave consent to be tested, 4 could not be contacted, and 1 did not give consent to testing. The study included the 10 adult individuals, aged 18-years-old and above, with symptoms of ataxia, and who tested positive for SCA2 gene. Individuals who did not give consent or did not undergo genetic testing were excluded. A chart review was conducted to assess the genotype-phenotype correlation including the clinical features of patients based on motor cerebellar manifestations, oculomotor disturbances, corticospinal tract dysfunction, extrapyramidal features, peripheral neuropathy manifestations, sleep disorders, cognitive decline and autonomic dysfunction. The SARA, with a total rating score of 30, was derived from the Ataxia Study Group (ASG) in Strasbourg, France, was used to assess ataxic features. Data was analyzed using JAMOVI software. Linear regression analyses were employed to examine the relationships between the SARA score, age of onset, duration of disease, and CAG repeat length. Pearson's correlation coefficients and Spearman's Rho coefficients were calculated. The statistical significance level was established for p-values < 0.05.
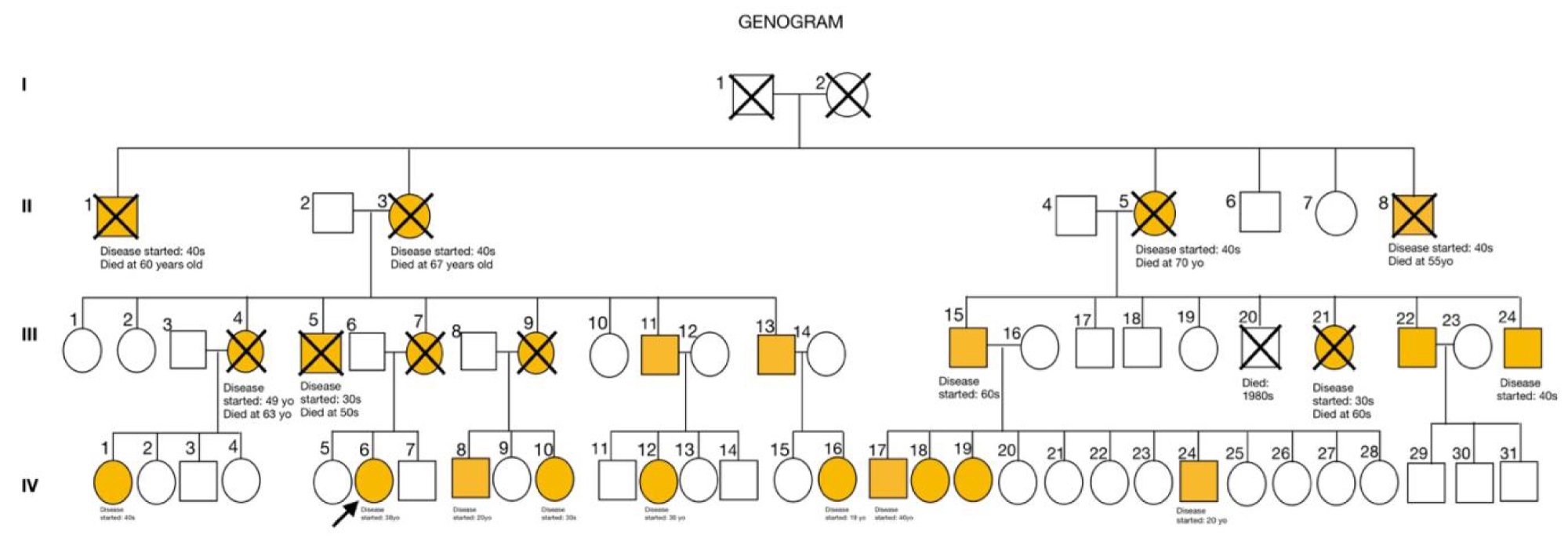 Figure 1: Genogram of proband (arrow) indicating symptomatic relatives (orange boxes and circles).
View Figure 1
Figure 1: Genogram of proband (arrow) indicating symptomatic relatives (orange boxes and circles).
View Figure 1
The average age included in the study is 46.8 ± 11.7 years, the age of onset ranged from 18 to 59 years of age (mean: 46.8 ± 11.7) and the mean disease duration is 10.2 ± 6.3 years. Male to female ratio of disease occurrence was 6:4 (Table 1).
Table 1: Demographics of 10 Filipino genetically-positive SCA2 patients (N = 10). View Table 1
Eight out of the 10 patients presented with gait ataxia as the initial. One patient (IV-16) first developed tremors of the left hand as the presenting symptom with rapid course of progression to dysarthria and gait ataxia. Another patient (IV-24) noted chronic low back pain and non-vertiginous dizziness as the initial symptom and later developed gait imbalance. Table 2 shows the clinical features of the patients, including motor cerebellar manifestations present in 70-100% of the patients.
Table 2: Clinical Features of 10 Filipino genetically-positive SCA2 patients (N = 10). View Table 2
All were heterozygous for the mutation, as evidenced by the presence of both normal and abnormal CAG repeat lengths (Table 1). The CAG repeat length for the group ranged from 39 to 50 repeats (mean: 42.3 ± 3.06). Nineteen repeats (mean: 21.4 ± 1.26) were present in only 2 individuals.
Only age at onset is significantly correlated with size of the CAG expansion (Figure 2A). There is a negative correlation between age of disease onset and number of CAG repeat units in the expanded allele (Pearson's r = -0.778, p-value 0.008; Spearman's rho = -0.932; p-value < 0.001). Linear regression analysis reveals that CAG repeat length accounts for about 60.5% of variance in age of onset (Table 3). Furthermore, duration of the disease measured in years and SARA is not significantly correlated with CAG repeat lengths (Figure 2B and Figure 2C).
Table 3: Correlation matrix of the age of onset (years), disease duration (years), and SARA scores with CAG repeat lengths. View Table 3
 Figure 2: Scatterplot diagrams (A) Showing a statistically significant inverse relationship between age of disease onset measured in years and number of CAG repeat length in the pathogenic allele in 10 SCA 2 patients. Pearson correlation coefficient r = -0.778 and Spearman's rho = -0.932 (p = 0.008 and p < 0.001 respectively). (B) Linear regression model showing no statistically significant correlation between disease duration measured in years and number of CAG repeat length in the pathogenic allele in 10 SCA 2 patients (Pearson correlation coefficient r = 0.259 and Spearman's rho = 0.299 (p = 0.469 and p = 0.401 respectively)) and (C) Between SARA scores and number of CAG repeat length in the pathogenic allele in 10 SCA 2 patients (Pearson correlation coefficient r = 0.558 and Spearman's rho = 0.197 (p = 0.094 and p = 0.586 respectively)).
View Figure 2
Figure 2: Scatterplot diagrams (A) Showing a statistically significant inverse relationship between age of disease onset measured in years and number of CAG repeat length in the pathogenic allele in 10 SCA 2 patients. Pearson correlation coefficient r = -0.778 and Spearman's rho = -0.932 (p = 0.008 and p < 0.001 respectively). (B) Linear regression model showing no statistically significant correlation between disease duration measured in years and number of CAG repeat length in the pathogenic allele in 10 SCA 2 patients (Pearson correlation coefficient r = 0.259 and Spearman's rho = 0.299 (p = 0.469 and p = 0.401 respectively)) and (C) Between SARA scores and number of CAG repeat length in the pathogenic allele in 10 SCA 2 patients (Pearson correlation coefficient r = 0.558 and Spearman's rho = 0.197 (p = 0.094 and p = 0.586 respectively)).
View Figure 2
There were no sporadic cases noted in this study. The average of age of onset of the group falls within the range frequently specified by literature. The average disease duration is also congruent with the range frequently indicated by studies on patients with SCA2 [2,6,16-18] and demonstrates that the disease has a slower rate of progression as compared to other types of SCA [12,13].
Notably, there is wide variability among the SARA scores, ranging from 0.5 to 30. There seems to be no significant correlation between the age of disease of onset and the severity of symptoms. This is demonstrated by two individuals (IV-10 and IV-12), who were noted to have relatively early ages of onset (30-years-old), but who also presented with symptoms of mild severity (SARA scores of 13 and 2, respectively). Similar findings were demonstrated by a study by Monte and researchers (2018), which showed that the progression rates of SARA were not constant during the long disease duration of SCA2 symptomatic patients and that the speed of progression of SARA scale was not uniform during the disease process and could vary according to the stage of the disease [19].
Additionally, the findings of this study confirmed the progressive, multisystemic and pleomorphic nature of SCA2 [2].
Interestingly, the most common clinical features noted in this study were cerebellar dysarthria present in all 10 patients, followed by gait ataxia, postural instability, then by parkinsonian signs such as rigidity, tremors, and bradykinesia seen in 20-30% of patients. Nonmotor symptoms were also noted for this population. Such symptoms include sleep disorders like primary insomnia, peripheral neuropathies, subjective cognitive decline, and even autonomic dysfunction like urinary incontinence present in one patient. Due to results should alert clinicians that the frequency of nonmotor symptoms may have previously been underestimated in SCA2.
In this study, an expanded CAG repeat at the SCA2 locus was found in the 10 patients included. All were heterozygous for the disease, and the size distribution of the normal SCA2 allele showed little polymorphism (range from 19-22, mean: 21.4 ± 1.26). Normal alleles have 31 or fewer CAG repeats, with 22 repeat alleles being present in more than 90% of normal individuals worldwide [16]. It is believed that the CAA interruptions of SCA2 normal alleles play a critical role in conferring stability to the CAG repeat expansion, and affect the secondary RNA structure, thus being likely contributors to phenotype variability [12]. However, it is unknown if these normal SCA2 repeats are interrupted, an observation usually made in other SCA2 normal alleles. This is one possible explanation for the variability in the clinical features noted. The individuals studied have relatively longer repeats than the common range (range 39-50, mean: 42.3 ± 3.06), which suggests that the disease was present for several generations. This is supported by findings from previous studies, which provide evidence of unstable intergeneration transmissions and anticipation [1,4,9].
The data of this study confirmed that (1) There is a significant correlation between expanded CAG repeats and age of onset, and (2) The expanded CAG size in the ATXN2 gene is responsible of the approximately 60.5% of the age at onset variability in patients. This suggests the existence of other genetic and non-genetic factors influencing the SCA2 age at onset [2,19]. It is noteworthy to emphasize that there is no significant correlation between the CAG repeat length and disease duration. The pathological threshold of CAG expansion is lower and the curve relating to CAG repeat number to age of onset is steeper than in other diseases caused by translated CAG expansions. The SCA2 protein seems, therefore, to be very sensitive to polyglutamine or to exert no protection against a putative polyglutamine toxicity [19].
Finally, this study suggests that there is no significant correlation between disease severity, as measured by SARA scores, and CAG repeat lengths. This is in accordance with previous studies on different types of spinocerebellar ataxias, which concluded that there is ambiguity regarding the diagnostic significance of the expansion length of CAG repeats [2]. It should be noted that this is a cross-sectional, retrospective research study, which can only reflect associations between variables at a given point in time. It would be prudent to investigate whether there is correlation between CAG repeat lengths and the progression of disease severity, reflected by changes in SARA scores over time. Although the study did confirm findings that were also present in other studies involving SCA2 in a different race [1,2,10,11,17,18] there is still some variability of results in terms of epidemiology, clinical features and disease duration. Additionally, studies investigating the complex nature of SCA2 should be done to elucidate further this disease and to explore treatment options for persons afflicted with this disease. This is the first study attempting to correlate the genotypic and phenotypic characteristic of spinocerebellar ataxia. There is no study regarding the prevalence of the disease in the country and there are no present published literature regarding the description of phenotype-genotype characteristics of different SCA types in the country.
In conclusion, there is sparsity of available literature regarding autosomal dominant spinocerebellar ataxias in Filipinos. This research reports the first clinical and molecular characterization of a Filipino family diagnosed with genetically-positive SCA2 gene. It should be noted that the study involved only a founder population and data tends to be more homogenous both genetically and environmentally. This may facilitate bias in terms of genotype analysis. Furthermore, findings in founder populations may be difficult to generalize to other populations. Hence, it is recommended to do a prospective study on a larger Filipino population with this disease. Prospective studies exploring the different domains of SARA and correlation with other ataxia rating score is recommended. The present study showed congruence to similar studies investigating the complex symptomatology of SCA2 and the data presented here can contribute to existing reports to better understand the natural history of brain degeneration in this genetic condition to pave way for the development of disease-modifying treatment.
This study would not have been possible without the support of the following physicians, Hiroshi Doi, MD, PhD and Shingo Ikeda, MD of Yokohama University in Japan, who provided expertise that greatly assisted the study and for sharing their pearls of wisdom during the course of this research. I am especially indebted to Dr. Eri Uematsu who have been supportive of this study from the beginning and who worked actively and provided extensive professional guidance in completing this research.
None.