Abstract
Dementia with Lewy bodies (DLB) and Parkinson's disease-dementia (PDD) are two closely related major neurocognitive disorders with Lewy bodies of unknown etiology, showing notable overlap in their clinical presentation, pathological features, biochemistry, and genetic risk factors. According to international consensus, their diagnosis is based on an arbitrary distinction between the time of onset of motor and cognitive symptoms: dementia preceding parkinsonism in DLB, while it develops after onset of parkinsonism in PDD (the one-year rule). Clinically, both syndromes show cognitive impairment with severe deficits in executive function, visuo-spatial processing, fluctuating attention and parkinsonism, with higher prevalence of Alzheimer-type lesions in DLB that may account for earlier onset and severity of cognitive deficits. These are also associated with multiple neurotransmitter deficits indicating that cognitive impairment in Lewy body disesses is multifactorial. Recent intra vitam neuroimaging, clinico-pathological studies and animal models suggest that DLB and PDD represent closely related but different, heterogenic subtypes of an α-synuclein-associated disease spectrum (Lewy body diseases) or parts of a continuum with PDD at the mild end of the spectrum, DLB in the middle, and DLB+AD at the more severe end. In view of the controversies about the nosology of both disorders, continuous effort is necessary to differentiate them more clearly and to clarify the underlying pathogenic mechanisms in order to enable effective mechanistic-based treatment, while, currently, no disease-modifying therapies are available.
Keywords
Dementia with Lewy bodies, Parkinson's disease-dementia, Synucleinopathies, Clinical features, Diagnostic criteria, Neuropathology, Pathogenesis, Management
Abbreviations
5HT: Serotonin, ACh: Acetylcholine, AChE: Acetylcholinesterase, AD: Alzheimer's Disease, αSyn: α-Synuclein, ChEI: Cholinesterase Inhibitor, CSF: Cerebrospinal Fluid, DAT: Dopamine Transporter, DLB: Dementia with Lewy Bodies, LB: Lewy Body, LC: Locus Ceruleus, MCI: Mild Cognitive Impairment, PD: Parkinson's Disease, PDD: Parkinson's Disease Dementia, SET: Serotonin Transporter
Background
Dementia with Lewy bodies (DLB) and Parkinson's disease dementia (PDD) are major neurocognitive disorders with α-synuclein (αSyn) deposits/Lewy bodies (LB), the nosological relations of which are under discussion (see [1,2]). Both entities are closely related with notable overlap in their clinical presentation, pathological features, biochemistry, and genetic risk factors. Even though the diagnostic criteria for these α-synucleinopathies are well established, their precise clinical diagnosis is often difficult, because DLB, Parkinson's disease (PD), PDD and Alzheimer's disease (AD) share clinical, morphological and biochemical characteristics, suggesting "overlap" syndromes between synucleinopathies (PD) and tauopathies (AD) [3]. Based on international consensus, DLB is diagnosed when dementia precedes parkinsonian signs for at least 1 year [4], whereas in PDD cognitive impairment develops in the setting of well-established PD [5]. The clinical features of both entities are similar and include parkinsonism with severe deficits in executive function, visuo-spatial processing, cognitive fluctuations, visual hallucinations, REM sleep behavior disorder (RBD), and memory disorders, the latter being more severe in PDD [6,7]. Despite different temporal sequences of motor and cognitive deficits (the 1 year rule), both syndromes show similar, albeit locally and quantitatively divergent morphological lesions, with variable mixtures of αSyn/LB and AD-related lesions, but increased cortical Aβ and tau load in DLB [8,9]. The multifold overlap between DLB and PDD has led to debating whether they should be classified as the same disease [10] or parts of a continuum reflecting a regional cerebral pathology [1] or distinct "diseases" with different predisposing genetic factors but sharing those with PD and AD [11]. However, recent studies indicate a regional overlap of multiple pathologies between synucleinopathies (PD) and tauopathies (AD) [3,12].
Clinical Manifestations and Diagnostic Criteria
Clinically, DLB is marked by four core symptoms: fluctuating cognition, parkinsonism, visual hallucinations, RBD, and cognitive impairment across multiple domains with moderate memory impairment [13]. DLB is a heterogenous disease. Cluster analysis yielded 3 different groups: cognitive-predominant (showing longer duration), neuropsychotic-dominant (older at onset), and parkinson-predominant (showing a shorter time from onset to presence of parkinsonism and dementia [14]). DLB patients with mild cognitive impairment (MCI) perform worse on visuospatial function and letter fluency tests and better on episodic memory tests than AD-MCI [15]. Cognitive impairment in PD patients being more common in the akinetic/rigid phenotype than in tremor-dominant and mixed phenotypes [16] is similar in quality to what is observed in DLB [6], but the rate of cognitive decline is faster in DLB than in PDD and AD [17,18], and shows more severely impaired frontal and temporal area-associated cognitive profiles [19]. The clinical overlaps and dissimilarities between DLB and PDD have been summarized recently (Table 1).
Table 1: Clinical overlap and dissimilarities between dementia with Lewy bodies (DLB) and Parkinson disease with dementia (PDD) [2].
View Table 1
DLB accounts for up to 5% of all dementia cases, while PDD affects between 75 and 95% of PD patients, with incidence rates of 2.5 to 3.5/100,000 person/years, the incidence of both syndromes being around 6% [20]. The average age at diagnosis is 70 to 76 years [21]. Individuals with DLB and PDD have increased mortality compared to the general population [22], the average survival time for DLB from the beginning of symptoms is 5-8 years, while PDD antedates death by about 4 years [23].
Genetics
Both DLB and PDD are primarily sporadic diseases, yet some different genetic factors may be involved, none of which, however, being diagnostic [24]. Glucocerebrosidase mutations and SNCA (αSyn gene) have been associated with both DLB and PDD [25-27]. In the clinical continuum between DLB and PDD, carriers of severe glucocerebrosidase mutations have a clinical phenotype that is closer to DLB [28]. APOE4 allele frequency is higher in DLB compared to PDD and may influence the risk for DLB [29]. Genome-wide association studies (GWAS) have found associations of variants of APOE, SNCA and other loci with DLB but not PDD, suggesting that DLB may share genetic loci with PD and AD [11], although the ways of involvement may be different, underpinning the concept of different pathomechanisms for PDD and DLB. However, the genetic differences between both entities have, so far, not been studied in detail [30].
Diagnostic Biomarkers
Multimodal imaging methods have improved ante mortem diagnosis of both DLB and PDD [31,32]. Striatal dopamine imaging shows reduced dopamine transporter (DAT) binding in putamen in both entities [33] indicating nigrostriatal loss being more severe in PDD consistent with the severity of parkinsonism, while others found no differences between DLB and PDD [34]. FDG-PET and 123I-FP-CIT SPECT are used to distinguish DLB from AD but have limited sensitivity since DAT imaging is also abnormal in other atypical parkinsonian syndromes [35]. Recent studies showed lower 123I-FP-CIT bindings to the striatal DAT, but not to the extrastriatal serotinin transporter (SET) in PD compared to DLB [36]. The sensitivity of FP-CIT imaging in probable MCI-DLB was 61% [37]. Semiquantitative ratings of FP-CIT SPECT scans were more accurate than visual ratings [38].
Cardiac neuroimaging using 123 I-metaiodobenzylguanidine (MIBG) as marker of postganglionic sympathetic innervation, shows reduced MIBG uptake in both DLB and PDD and allows no differentiation between the two disorders [39]. Its diagnostic usefulness in early stages of DLB was suggested [40].
So far, the contribution of MRI to the diagnosis of DLB and PDD is limited. Magnetic resonance volumetry (VBM), shows cortical thinning in different cortical areas but similar relative preservation of the medial temporal lobe (MTL) in both disorders. There is similar atrophy of striopallidum but more extensive atrophy of thalamus in DLB. The latter may be a marker of disease severity indicating rapid decline [41], while evaluation of white matter hypointensities and MTL atrophy may indicate progression of AD-related pathology in DLB and perhaps can distinguish DLB from PDD [42,43].
Functional MRI showed a different disruption of cortical functional connectivity in PDD (predominant frontal disruption) compared to DLB (predominant parietal and occipital) [44]. Molecular changes in the pulvinar may result in decreased cortical synchrony in DLB [45]. Brain PET revealed occipital hypoperfusion in both DLB and PDD with no or only subtle metabolic differences [46,47]. FDG PET is one alternative biomarker that can differentiate AD and DLB but lacks the evidence base of both DAT and MIBG scans [48]. Mapping of brain acetylcholinesterase (ACHE) showed alterations in cortical and subcortical levels in both PDD and DLB [49,50]. Recent PET studies of the vesicular ACh transporter (VAChT) showed more extensive reductions affecting neocortical, limbic and thalamic regions in DLB than in AD [51]. 11C PIB-PET imaging showed more frequent and severe Aβ brain deposition in DLB than in PDD [52,53], predictive of faster degeneration in cortex and striatum [54]. Tau pathology visialized by PET imaging along with temporal atrophy, indicative of coexisting AD pathology, is more common in DLB compared to PDD [33], amyloid playing an important role for tau accumulation [55]. A recent review of neuroimaging in DLB emphasized the importance of a multimodel approach [56].
Quantitative EEG features may specifically differentiate DLB, PDD and AD [57], and transcranial sonographic hyperechogenicity of substantia nigra is more common in DLB tha in PDD [58].
Many cerebrospinal fluid (CSF) and some plasma biomarkers have been identified in DLB and PDD, but very few studies examined samples from both disorders simultaneously, and only a minority have been confirmed by postmortem studies [59,60]. Large scale studies showed lower levels of Aβ42 and higher tau in DLB than in PDD. A CSF AD profile with elevated Aβ40 but decreased Aβ42 and higher tau levels is more common in DLB than in PDD [61], which may be related to increased AD pathology. Levels of αSyn oligomers are increased in PDD but not in DLB [59,62,63]. However, the current use of CSF biomarkers in the diagnosis of DLB and PDD is not yet recommended by the American Academy of Neurology [64].
Neuropathological Features
Both DLB and PDD show similar morphological features, with a variable mixture of αSyn/LB and AD-related lesions and a multi-organ distribution of αSyn pathology [2,65]. A common pathophysiological factor is synaptic dysfunction due to initial aggregation of αSyn in presynapses causing functional disconnection [66,67] due to interference with axonal transport and neurotransmitter deprivation [68]. Studies of large cohorts have shown a strong correlation between both cortical Lewy and AD-related pathologies, suggesting that phosphorylated αSyn promotes the phosphorylation of tau [69,70], while tau oligomers mediate αSyn toxicity [71]. The relationship between phosphorylated αSyn and tau accumulation to Aβ deposition in cerebral cortex supports an overlap in the pathology of DLB, PDD and AD and that Aβ promotes the accumulation of both αSyn and tau [3,12,72]. A recent study showed that cerebral tau, Aβ and αSyn pathologies are strong predictors of a shorter interval between onset of motor and cognitive symptoms and shorter survival in LB dementias [73]. Thus, cognitive impairment in both DLB and PDD is not only induced by αSyn caused neurodegeneration but by multiple regional pathological scores. Despite many similiarities, several morphological differences have been demonstrated, e.g. higher amyloid load in striatum [74,75] and amygdala, higher Aβ phases and neuritic plaque scores in cortex [76], as well as more frequent tau pathology in striatum in DLB [77]. Minor differences are more severe αSyn load in hippocampal subareas C2/3 in DLB and differences in the severity and distribution pattern of substantia nigra lesions (predominant neuronal loss in ventrolateral vs. dorsolateral cell groups in PDD vs. DLB) [78,79]. Recent morphological studies revealed differential vulnerability of the anterior insular cortex to αSyn pathology in PDD and DLB due to a decreasing gradient of αSyn immunoreactivity from the anterior allocortical to the posterior isocortical granular insula in iLBD, PD, PDD, and DLB [80] (Table 2 [2]). The heterogeneous neurochemistry of both entities also supports the overlap between synucleinopathies and tauopathies [3].
Table 2: Morphological overlap and dissimilarities between DLB and PDD [2].
View Table 2
At present, neuropathological diagnosis of PDD and DLB without sufficient clinical data would be difficult due to the heterogeneity of synucleinopathies, but preliminary criteria have been proposed (Table 3). For their validation, further clinicopathological studies will be necessary in order to further promote our understanding of the molecuar and pathogenic backgrounds of both DLB and PDD.
Table 3: Preliminary neuropathological criteria for dementia with Lewy bodies (DLB) and Parkinson disease with dementia (PDD) [2].
View Table 3
Pathophysiology of Cognitive Impairment
The neurobiological basis for cognitive impairment in DLB and PDD is multifocal, related to a synergistic effect of both αSyn/LB and AD pathologies and dysfunction of dopaminergic, noradrenalinergic, serotonergic, and cholinergic systems [70,81,82]. The emergence of PDD and DLB occurs on the background of severe dopamine deficits and correlates with a marked loss of limbic and cortically projecting dopamine, noradreanaline, serotonin, and ACh neurons. The relationship between these lesions is not yet fully understood.
Cognitive deficits, already seen in early PD are associated with striatal and extrastriatal dopamine deficiency [83] which results in abnormal processing in the frontostriatal circuit with reduced prefrontal and parietal metabolism [84] and in the salience network of the medial temporal lobe [85], whereas mesocortical dopaminergic transmission is less involved [86]. However, dopamine deficiency in PDD and DLB have been shown to impair a wide network of brain areas [87]. Reduced striatal DAT binding in DLB as evidence for compromised dopaminergic neurotransmission also indicates significant cholinergic deficit [88]. Cortical cholinergic denervation associated with cognitive impairment occurs in the context of significant caudate dopaminergic denervation, indicating interactive contribution of both systems to cognitive impairment in PDD and DLB [89]. Reduction of cholinergic markers in both PDD and DLB [49,50] is due to early degeneration of the corticopetal basal forebrain cholinergic projection involving both the nucleus basalis of Meynert and the nucleus of the diagonal band of Broca [90-93]. Pedunculopontine cholinergic cell loss is seen in hallucinating PDD patients but not in DLB patients [94], and is also relatively spared in AD [95]. Patterns of acetylcholine (ACh) deterioration are different in DLB from those in AD, with synergistic actions between Aβ and αSyn causing additional symptoms that accelerate the disease course [96]. On the other hand, galanin upregulation within the basal forebrain cholinergic system in DLB, similar to that in AD but not in PDD, may represent an intrinsic adaptive response to neurodegeneration of these areas [97]. Indices of reduced cortical cholinergic innervation in DLB are similar to PDD and lower than in AD [98]. Cortical cholinergic acitivity was lower in hallucinating compared to nonhallucinating DLB cases, while SET activity was relatively preserved [99].
Severe pathology also involves the noradrenergic locus ceruleus (LC) [100] and the serotonergic dorsal raphe nucleus [101] as well as the ventral tegmental area not always associated with coincidental AD lesions [102]. LC neuronal loss and the accompanying norepinephrinergic deficiency are an important cause and pharmacological target for the (symptomatic) treatment of PD/PDD/DLB [103,104]. The prominent role of serotonergic degeneration also involving the anterior caudate nucleus, the orbitofrontal and cingulate cortex for neuropsychiatric symptoms in PD [105], emphasizes its important role in both PDD and DLB, and stimulates new insight into novel treatments by modulating 5-HT receptors [106].
Whereas dopamine dysfunction has been highlighted because of its obvious role in PD/PDD/DLB, the role of the other neurotransmitter systems in the pathogenesis of cognitive impairment in these LB disorders has not yet been sufficiently explored, but its close relationship with combined LB and AD-pathologies suggests synergism of variable pathogenic mechanisms in the clinical manifestation of these disorders [82].
Pathogenesis
The clinicopathological features of DLB, PDD and other synucleinopathies are highly variable and heterogenous [107], documented by 4 current staging systems in use for LB disorders, one for PD [108,109], another one for DLB [110], revised guidelines [13,111,112], and a recent one [68]. Based on semiquantitative assessment of LBs, a staging of the chronological spread of pathology was proposed to designate its predictable caudorostal spreading [113], which, however, is not identical with the spreading of αSyn pathology [114]. The McKeith criteria distinguish between three types: (1) Brainstem-predominant LB pathology corresponding to classical PD with low probability of developing clinical dementia; (2) Limbic or transitional type (DLB), in which clinical dementia may be associated with severe AD-related pathologies, and (3) Diffuse neocortical LB pathology (Braak LB stages 5 and 6), strongly associated with clinical dementia. These criteria differ from Braak LB stages insofar as they do not strictly postulate a stepwise progression of αSyn pathology from the olfactory bulb and medulla to midbrain, limbic areas and neocortex correlating with clinical dementia [115], since cases may show severe neocortical αSyn pathology with only minimal involvement of brainstem regions [116]. Moreover, this differs from the changes in DLB, where parkinsonism develops an average of 2 years after the onset of cognitive symptoms with a reported prevalence of parkinsonism between 66% and 92% [117].
Transcellular propagation of protein aggregate "seeds" has been proposed to mediate the progression of neurodegeneration in tauopathies and synucleinopathies [118,119]. This "prion-like" transmission of αSyn and other pathological proteins, appears also essential for the pathogenesis of both PDD and DLB [115,119-121]. It has been suggested that distinct species of αSyn are responsible for propagation and differences of regional distribution of lesions in various synucleinopahies [122], and that different strains of pathological αSyn are involved in the heterogeneity of synucleinopathies [123]. This has been confirmed by recent studies demonstrating that distinct αSyn strains are determined by both misfolded seeds and intracellular environments [124] and that different types of αSyn assemblies have a unique and causative role in distinct synucleinopathies [125-127].
Management
Currently, no disease-modifying therapies of LB dementias are available. Clinical management of both disorders include cholinesterase inhibitors (ChEIs) to treat cognitive and psychiatric symptoms [128,129]. Although the effects of ChEIs were relatively small [130], they gave a better response of cognitive impairment in DLB and PDD than in AD [131]. The use of antipsychotics should be avoided given the risk of serious reactions in DLB [13], while atypical antipsychotic agents like quetiapine and clozapine less likely may exacerbate parkinsonism. Levodopa was generally well tolerated, but produced less motor response in DLB than in PDD [132] and may increase the risk of psychosis [128]. Bilateral deep brain stimulation of the nucleus basalis of Meynert for PDD showed no improvement in primary cognitive outcomes [133]. A recent review of nonpharmacological interventions did not offer any definite recommendations [134]. Future therapeutic strategies might include disease-modifying methods, based on vaccination trials against αSyn, Aβ and tau proteins [135,136], inhibition of αSyn aggregation or promoting degeneration, prevention of cell-to-cell transmission of pathological αSyn, deep brain stimulation of the cholinergic nucleus basalis of Meynert or transcranial current stimulation [137,138]. Preliminary results of anti-αSyn-immunotherapy in a combined model of synucleinopathy [139] may open the way to potential new treatments.
Conclusion and Future Outlook
DLB a d PDD are multi-faced neurocognitive disorders with LB and AD-related pathologies, sharing many clinical, genetic, morphological, histochemical, neuroimaging, and pathogenetical features. Up to now, a clear and definie destinction between the two entities other than the arbitrary timing of the appearance of motor and cognitive impairments (1-year rule) has not been achieved [4,13], and the recent Movement Disorder Society (MDS) Panel has rather confused than clarified the distinction between the two disorders [140]. In view of the clinical and neuropathological heterogeneity of both disorders that share a similar if not identical pathogenesis and pathophsiology, the question whether the 1-year rule may be a biologically valid distinction or whether they are subtypes of a continuum of LB disorders awaits further elucidation. Despite validitated criteria for DLB and PDD [5,13,141], in almost 50% of the cases, the clinical diagnosis is incorrect [142,143], whereas the specificity was below 60% [144]. The sex ratio in DLB is balanced between AD and PDD and, thus, also suggests that DLB is a distinct disease with characteristics intermediate between AD and PD [145], while recent MRI findings indicate that DLB is more similar to AD than to PDD [146].
The available clinical and morphological data clearly indicate that in DLB, the topograpical spreading pattern of αSyn/LB pathology differs from that of PDD indicating pathogenic differences between them [147]. Despite considerable overlap between both conditions, recent studies have demonstrated differences in the quantity and distribution pattern of both αSyn and AD-related pathologies, with higher Aβ and tau load in cortical and subcortical areas in DLB, indicating an overlap between DLB and AD (DLB+AD). This suggests a Lewy body disease continuum with PD non-demented and PDD at the mild end of the spectrum, DLB in the middle, and DLB+AD at the more severe end (Figure 1). Interaction between αSyn, Aβ and tau may represent a basic mechanism in the pathogenesis of PDD/DLB and AD, leading to neurodegeneration depending on genetic backgrounds or environmental factors [148]. Further elucidation of the relations between DLB and PDD including better insight into common genetic risk factors and pathogenetic molecular pathways responsible for the clinical manifestation of both disorders will be necessary as the basis for future preventive and cognitive treatment options.
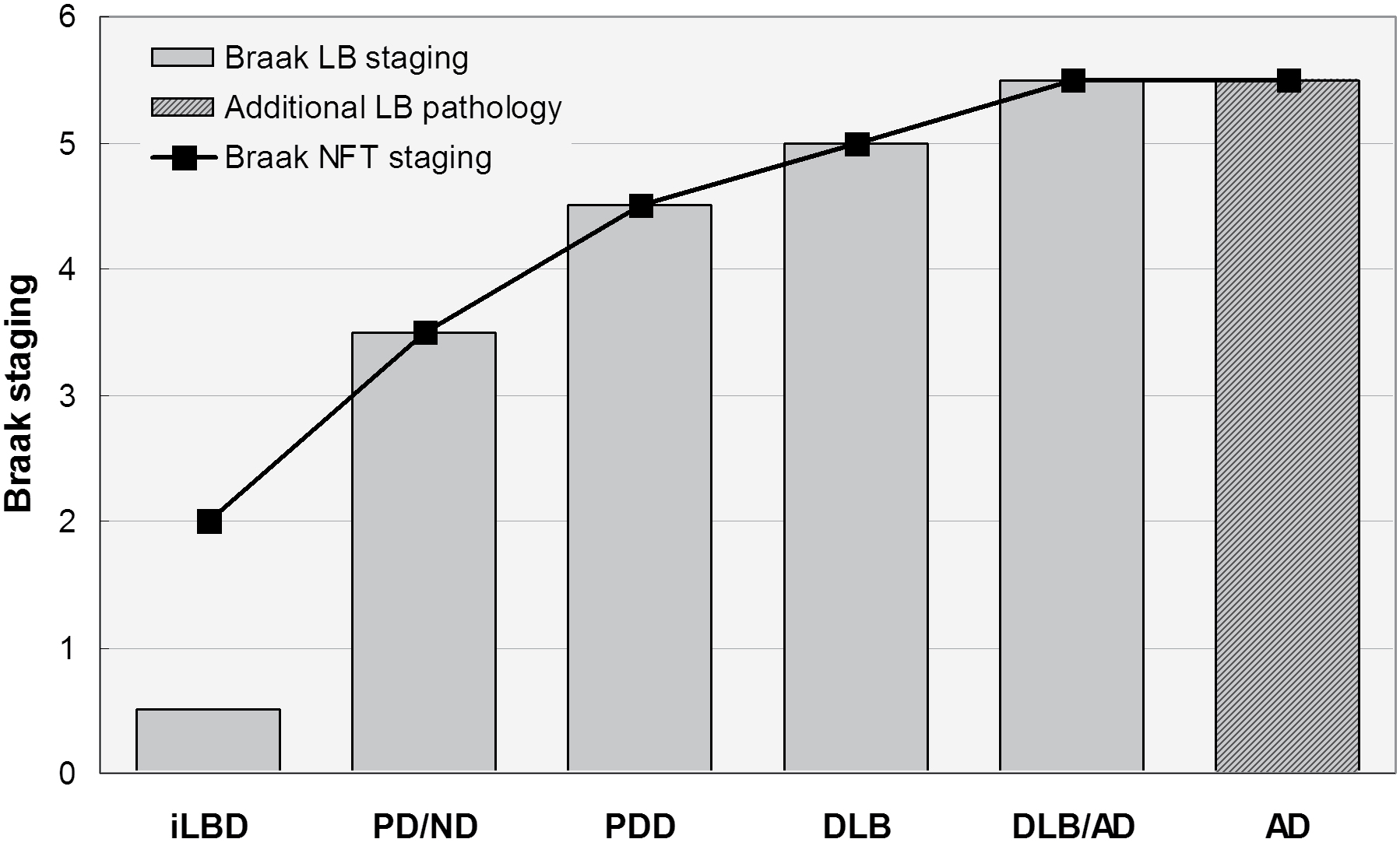 Figure 1: Spectrum of Lewy body diseases with relations between Braak LB and Braak neuritic (NFT) stages ranging from iLBD to DLB/AD. Modified from [2].
Figure 1: Spectrum of Lewy body diseases with relations between Braak LB and Braak neuritic (NFT) stages ranging from iLBD to DLB/AD. Modified from [2].
LB: Lewy Body; NFT: Neurofibrillary Tangle; iLBD: Incidental Lewy Body Disease; PD/ND: Parkinson Disease - Not Demented; PDD: Parkinson's Disease-Dementia; DLB: Dementia with Lewy Bodies; DLB/AD: DLB with Alzheimer Disease; AD Alzheimer's Disease.
View Figure 1
Core tip
Dementia with Lewy bodies (DLB) and Parkinson's disease-dementia (PDD) are two closely related neurocognitive disorders with overlap in their clinical, morphological and pathogenic features. Their diagnosis is based on an arbitrary distinction between the time of onset of motor and cognitive symptoms ("one-year rule") related to a variable mixture of Lewy body and Alzheimer-related pathologies, and multiple neurotransmitter deficits. DLB and PDD are suggested to represent subtypes of an α-synuclein-associated disease spectrum (Lewy body diseases) or parts of a continuum showing strong pathogenic correlations with Alzheimer's disease. The nosology of both disorders is a matter of discussion, their differential diagnosis can be challenging and much effort is necessary to differentiate them more clearly and to clarify the underlying pathogenic mechanisms to enable effective treatment, while, currently, no disease-modifying therapies are available.
Conflict-of-Interest Statement
The author has no conflict of interest to declare.
Acknowledgement
The study was supported by the Society for Support of Research in Experimental Neurology, Vienna, Austria.
References
-
Jellinger KA, Korczyn AD (2018) Are dementia with Lewy bodies and Parkinson's disease dementia the same disease? BMC Med 16: 34.
-
Jellinger KA (2018) Dementia with Lewy bodies and Parkinson's disease-dementia: current concepts and controversies. J Neural Transm (Vienna) 125: 615-650.
-
Foguem C, Manckoundia P (2018) Lewy body disease: Clinical and pathological "overlap syndrome" between synucleinopathies (Parkinson disease) and tauopathies (Alzheimer disease). Curr Neurol Neurosci Rep 18: 24.
-
McKeith IG, Dickson DW, Lowe J, Emre M, O'Brien JT, et al. (2005) Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65: 1863-1872.
-
Emre M, Aarsland D, Brown R, Burn DJ, Duyckaerts C, et al. (2007) Clinical diagnostic criteria for dementia associated with Parkinson's disease. Mov Disord 22: 1689-1707.
-
Aldridge GM, Birnschein A, Denburg NL, Narayanan NS (2018) Parkinson's disease dementia and dementia with Lewy bodies have similar neuropsychological profiles. Front Neurol 9: 123.
-
Lippa CF, Duda JE, Grossman M, Hurtig HI, Aarsland D, et al. (2007) DLB and PDD boundary issues: diagnosis, treatment, molecular pathology, and biomarkers. Neurology 68: 812-819.
-
Garcia-Esparcia P, Lopez-Gonzalez I, Grau-Rivera O, Garcia-Garrido MF, Konetti A, et al. (2017) Dementia with Lewy bodies: molecular pathology in the frontal cortex in typical and rapidly progressive forms. Front Neurol 8: 89.
-
Walker Z, Moreno E, Thomas A, Inglis F, Tabet N, et al. (2015) Clinical usefulness of dopamine transporter SPECT imaging with 123I-FP-CIT in patients with possible dementia with Lewy bodies: randomised study. Br J Psychiatry 206: 145-152.
-
Friedman JH (2018) Dementia with Lewy bodies and Parkinson's disease dementia become the same disease. Parkinsonism Relat Disord 46: S6-S9.
-
Walton RL, Soto-Ortolaza AI, Murray ME, Lorenzo-Betancor O, Ogaki K, et al. (2016) TREM2 p.R47H substitution is not associated with dementia with Lewy bodies. Neurol Genet 2: e85.
-
Colom-Cadena M, Grau-Rivera O, Planellas L, Cerquera C, Morenas E, et al. (2017) Regional overlap of pathologies in Lewy body disorders. J Neuropathol Exp Neurol 76: 216-224.
-
McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor JP, et al. (2017) Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 89: 88-100.
-
Morenas-Rodriguez E, Sala I, Subirana A, Pascual-Goni E, Sanchez-Saudinos MB, et al. (2018) Clinical subtypes of dementia with Lewy bodies based on the initial clinical presentation. J Alzheimers Dis 64: 505-513.
-
Sadiq D, Whitfield T, Lee L, Stevens T, Costafreda S, et al. (2017) Prodromal dementia with Lewy bodies and prodromal Alzheimer's disease: a comparison of the cognitive and clinical profiles. J Alzheimers Dis 58: 463-470.
-
Svenningsson P, Westman E, Ballard C, Aarsland D (2012) Cognitive impairment in patients with Parkinson's disease: diagnosis, biomarkers, and treatment. Lancet Neurol 11: 697-707.
-
Blanc F, Mahmoudi R, Jonveaux T, Galmiche J, Chopard G, et al. (2017) Long-term cognitive outcome of Alzheimer's disease and dementia with Lewy bodies: dual disease is worse. Alzheimers Res Ther 9: 47.
-
Kramberger MG, Auestad B, Garcia-Ptacek S, Abdelnour C, Olmo JG, et al. (2017) Long-term cognitive decline in dementia with Lewy bodies in a large multicenter, international cohort. J Alzheimers Dis 57: 787-795.
-
Lee JE, Park HJ, Park B, Song SK, Sohn YH, et al. (2010) A comparative analysis of cognitive profiles and white-matter alterations using voxel-based diffusion tensor imaging between patients with Parkinson's disease dementia and dementia with Lewy bodies. J Neurol Neurosurg Psychiatry 81: 320-326.
-
Kane JPM, Surendranathan A, Bentley A, Barker SAH, Taylor JP, et al. (2018) Clinical prevalence of Lewy body dementia. Alzheimers Res Ther 10: 19.
-
Yang SK, Chen W, Su CH, Liu CH (2018) Incidence and comorbidity of dementia with Lewy bodies: a population-based cohort study. Behav Neurol 2018: 7631951.
-
Savica R, Grossardt BR, Bower JH, Ahlskog JE, Boeve BF, et al. (2017) Survival and causes of death among people with clinically diagnosed synucleinopathies with parkinsonism: a population-based study. JAMA Neurol 74: 839-846.
-
Kempster PA, O'Sullivan SS, Holton JL, Revesz T, Lees AJ (2010) Relationships between age and late progression of Parkinson's disease: a clinico-pathological study. Brain 133: 1755-1762.
-
Nussbaum RL (2018) Genetics of synucleinopathies. Cold Spring Harb Perspect Med 8.
-
Guella I, Evans DM, Szu-Tu C, Nosova E, Bortnick SF, et al. (2016) alpha-synuclein genetic variability: A biomarker for dementia in Parkinson disease. Ann Neurol 79: 991-999.
-
Schneider SA, Alcalay RN (2017) Neuropathology of genetic synucleinopathies with parkinsonism: Review of the literature. Mov Disord 32: 1504-1523.
-
Liu S, Wang XD, Wang Y, Shi Z, Cai L, et al. (2017) Clinical and neuroimaging characteristics of Chinese dementia with Lewy bodies. PLoS One 12: e0171802.
-
Cilia R, Tunesi S, Marotta G, Cereda E, Siri C, et al. (2016) Survival and dementia in GBA-associated Parkinson's disease: The mutation matters. Ann Neurol 80: 662-673.
-
Vergouw LJM, van Steenoven I, van de Berg WDJ, Teunissen CE, van Swieten JC, et al. (2017) An update on the genetics of dementia with Lewy bodies. Parkinsonism Relat Disord 43: 1-8.
-
Weil RS, Lashley TL, Bras J, Schrag AE, Schott JM (2017) Current concepts and controversies in the pathogenesis of Parkinson's disease dementia and Dementia with Lewy Bodies. F1000Res 6: 1604.
-
Saeed U, Compagnone J, Aviv RI, Strafella AP, Black SE, et al. (2017) Imaging biomarkers in Parkinson's disease and Parkinsonian syndromes: current and emerging concepts. Transl Neurodegener 6: 8.
-
Lehericy S, Vaillancourt DE, Seppi K, Monchi O, Rektorova I, et al. (2017) The role of high-field magnetic resonance imaging in parkinsonian disorders: Pushing the boundaries forward. Mov Disord 32: 510-525.
-
Gomperts SN, Marquie M, Locascio JJ, Bayer S, Johnson KA, et al. (2016) PET radioligands reveal the basis of dementia in Parkinson's disease and dementia with Lewy bodies. Neurodegener Dis 16: 118-124.
-
Kasanuki K, Heckman MG, Diehl NN, Murray ME, Koga S, et al. (2017) Regional analysis and genetic association of nigrostriatal degeneration in Lewy body disease. Mov Disord 32: 1584-1593.
-
Walker Z, Costa DC, Walker RW, Shaw K, Gacinovic S, et al. (2002) Differentiation of dementia with Lewy bodies from Alzheimer's disease using a dopaminergic presynaptic ligand. J Neurol Neurosurg Psychiatry 73: 134-140.
-
Joling M, Vriend C, van der Zande JJ, Lemstra AW, van den Heuvel OA, et al. (2018) Lower (123)I-FP-CIT binding to the striatal dopamine transporter, but not to the extrastriatal serotonin transporter, in Parkinson's disease compared with dementia with Lewy bodies. Neuroimage Clin 19: 130-136.
-
Thomas AJ, Donaghy P, Roberts G, Colloby SJ, Barnett NA, et al. (2018) Diagnostic accuracy of dopaminergic imaging in prodromal dementia with Lewy bodies. Psychol Med: 1-7.
-
McCleery J, Morgan S, Bradley KM, Noel-Storr AH, Ansorge O, et al. (2015) Dopamine transporter imaging for the diagnosis of dementia with Lewy bodies. Cochrane Database Syst Rev 1.
-
Goldstein DS, Sharabi Y (2017) The heart of PD: Lewy body diseases as neurocardiologic disorders. Brain Res.
-
Komatsu J, Samuraki M, Nakajima K, Arai H, Arai T, et al. (2018) (123) I-MIBG myocardial scintigraphy for the diagnosis of DLB: a multicentre 3-year follow-up study. J Neurol Neurosurg Psychiatry.
-
Watson R, Colloby SJ, Blamire AM, Wesnes KA, Wood J, et al. (2017) Does attentional dysfunction and thalamic atrophy predict decline in dementia with Lewy bodies? Parkinsonism Relat Disord 45: 69-74.
-
Joki H, Higashiyama Y, Nakae Y, Kugimoto C, Doi H, et al. (2018) White matter hyperintensities on MRI in dementia with Lewy bodies, Parkinson's disease with dementia, and Alzheimer's disease. J Neurol Sci 385: 99-104.
-
Elder GJ, Mactier K, Colloby SJ, Watson R, Blamire AM, et al. (2017) The influence of hippocampal atrophy on the cognitive phenotype of dementia with Lewy bodies. Int J Geriatr Psychiatry 32: 1182-1189.
-
Borroni B, Premi E, Formenti A, Turrone R, Alberici A, et al. (2015) Structural and functional imaging study in dementia with Lewy bodies and Parkinson's disease dementia. Parkinsonism Relat Disord 21: 1049-1055.
-
Erskine D, Ding J, Thomas AJ, Kaganovich A, Khundakar AA, et al. (2018) Molecular changes in the absence of severe pathology in the pulvinar in dementia with Lewy bodies. Mov Disord 33: 982-991.
-
Meyer PT, Frings L, Rucker G, Hellwig S (2017) 18F-FDG PET in Parkinsonism: Differential Diagnosis and Cognitive Impairment in Parkinson's disease. J Nucl Med 58: 1888-1898.
-
Whitwell JL, Graff-Radford J, Singh TD, Drubach DA, Senjem ML, et al. (2017) 18F-FDG PET in posterior cortical atrophy and dementia with Lewy bodies. J Nucl Med 58: 632-638.
-
Surendranathan A, O'Brien JT (2018) Clinical imaging in dementia with Lewy bodies. Evid Based Ment Health 21: 61-65.
-
Mazere J, Lamare F, Allard M, Fernandez P, Mayo W (2017) 123I-Iodobenzovesamicol SPECT imaging of cholinergic systems in dementia with Lewy bodies. J Nucl Med 58: 123-128.
-
Klein JC, Eggers C, Kalbe E, Weisenbach S, Hohmann C, et al. (2010) Neurotransmitter changes in dementia with Lewy bodies and Parkinson disease dementia in vivo. Neurology 74: 885-892.
-
Nejad-Davarani S, Koeppe RA, Albin RL, Frey KA, Muller M, et al. (2018) Quantification of brain cholinergic denervation in dementia with Lewy bodies using PET imaging with [(18)F]-FEOBV. Mol Psychiatry.
-
Donaghy PC, Firbank MJ, Thomas AJ, Lloyd J, Petrides G, et al. (2018) Clinical and imaging correlates of amyloid deposition in dementia with Lewy bodies. Mov Disord 33: 1130-1138.
-
Petrou M, Dwamena BA, Foerster BR, MacEachern MP, Bohnen NI, et al. (2015) Amyloid deposition in Parkinson's disease and cognitive impairment: a systematic review. Mov Disord 30: 928-935.
-
Sarro L, Senjem ML, Lundt ES, Przybelski SA, Lesnick TG, et al. (2016) Amyloid-beta deposition and regional grey matter atrophy rates in dementia with Lewy bodies. Brain 139: 2740-2750.
-
Lee SH, Cho H, Choi JY, Lee JH, Ryu YH, et al. (2017) Distinct patterns of amyloid-dependent tau accumulation in Lewy body diseases. Mov Disord 33: 262-272.
-
Yousaf T, Dervenoulas G, Valkimadi PE, Politis M (2018) Neuroimaging in Lewy body dementia. J Neurol.
-
Garn H, Coronel C, Waser M, Caravias G, Ransmayr G (2017) Differential diagnosis between patients with probable Alzheimer's disease, Parkinson's disease dementia, or dementia with Lewy bodies and frontotemporal dementia, behavioral variant, using quantitative electroencephalographic features. J Neural Transm (Vienna) 124: 569-581.
-
Favaretto S, Walter U, Baracchini C, Pompanin S, Busse C, et al. (2016) Accuracy of transcranial brain parenchyma sonography in the diagnosis of dementia with Lewy bodies. Eur J Neurol 23: 1322-1328.
-
Stuendl A, Kunadt M, Kruse N, Bartels C, Moebius W, et al. (2016) Induction of alpha-synuclein aggregate formation by CSF exosomes from patients with Parkinson's disease and dementia with Lewy bodies. Brain 139: 481-494.
-
Eusebi P, Giannandrea D, Biscetti L, Abraha I, Chiasserini D, et al. (2017) Diagnostic utility of cerebrospinal fluid alpha-synuclein in Parkinson's disease: A systematic review and meta-analysis. Mov Disord 32: 1389-1400.
-
van Steenoven I, Aarsland D, Weintraub D, Londos E, Blanc F, et al. (2016) Cerebrospinal fluid Alzheimer's disease biomarkers across the spectrum of Lewy body diseases: results from a large multicenter cohort. J Alzheimers Dis 54: 287-295.
-
Hansson O, Hall S, Ohrfelt A, Zetterberg H, Blennow K, et al. (2014) Levels of cerebrospinal fluid alpha-synuclein oligomers are increased in Parkinson's disease with dementia and dementia with Lewy bodies compared to Alzheimer's disease. Alzheimers Res Ther 6: 25.
-
Simonsen AH, Kuiperij B, El-Agnaf OM, Engelborghs S, Herukka SK, et al. (2016) The utility of alpha-synuclein as biofluid marker in neurodegenerative diseases: a systematic review of the literature. Biomark Med 10: 19-34.
-
Andersson M, Zetterberg H, Minthon L, Blennow K, Londos E (2011) The cognitive profile and CSF biomarkers in dementia with Lewy bodies and Parkinson's disease dementia. Int J Geriatr Psychiatry 26: 100-105.
-
Wakabayashi K, Miki Y (2018) [Multi-organ distribution of alpha-synuclein pathology in dementia with Lewy bodies]. Brain Nerve 70: 489-500.
-
Bellucci A, Mercuri NB, Venneri A, Faustini G, Longhena F, et al. (2016) Review: Parkinson's disease: from synaptic loss to connectome dysfunction. Neuropathol Appl Neurobiol 42: 77-94.
-
Schirinzi T, Madeo G, Martella G, Maltese M, Picconi B, et al. (2016) Early synaptic dysfunction in Parkinson's disease: Insights from animal models. Mov Disord 31: 802-813.
-
Uchihara T (2017) An order in Lewy body disorders: Retrograde degeneration in hyperbranching axons as a fundamental structural template accounting for focal/multifocal Lewy body disease. Neuropathology 37: 129-149.
-
Yan X, Uronen RL, Huttunen HJ (2018) The interaction of alpha-synuclein and tau: A molecular conspiracy in neurodegeneration? Semin Cell Dev Biol.
-
Irwin DJ, Lee VM, Trojanowski JQ (2013) Parkinson's disease dementia: convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat Rev Neurosci 14: 626-636.
-
Gerson JE, Farmer KM, Henson N, Castillo-Carranza DL, Carretero Murillo M, et al. (2018) Tau oligomers mediate alpha-synuclein toxicity and can be targeted by immunotherapy. Mol Neurodegener 15: 13.
-
Colom-Cadena M, Pegueroles J, Herrmann AG, Henstridge CM, Munoz L, et al. (2017) Synaptic phosphorylated alpha-synuclein in dementia with Lewy bodies. Brain 140: 3204-3214.
-
Irwin DJ, Grossman M, Weintraub D, Hurtig HI, Duda JE, et al. (2017) Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet Neurol 16: 55-65.
-
Jellinger KA, Attems J (2006) Does striatal pathology distinguish Parkinson disease with dementia and dementia with Lewy bodies? Acta Neuropathol 112: 253-260.
-
Halliday GM, Song YJ, Harding AJ (2011) Striatal beta-amyloid in dementia with Lewy bodies but not Parkinson's disease. J Neural Transm 118: 713-719.
-
Ruffmann C, Calboli FC, Bravi I, Gveric D, Curry LK, et al. (2016) Cortical Lewy bodies and Abeta burden are associated with prevalence and timing of dementia in Lewy body diseases. Neuropathol Appl Neurobiol 42: 436-450.
-
Hepp DH, Vergoossen DL, Huisman E, Lemstra AW, Berendse HW, et al. (2016) Distribution and load of amyloid-beta pathology in Parkinson disease and dementia with Lewy bodies. J Neuropathol Exp Neurol 75: 936-945.
-
Dickson DW, Braak H, Duda JE, Duyckaerts C, Gasser T, et al. (2009) Neuropathological assessment of Parkinson's disease: refining the diagnostic criteria. Lancet Neurol 8: 1150-1157.
-
Jellinger KA (2006) Pathological substrate of dementia in Parkinson's disease--its relation to DLB and DLBD. Parkinsonism Relat Disord 12: 119-120.
-
Fathy YY, Jonker AJ, Oudejans E, de Jong FJJ, van Dam AW, et al. (2018) Differential insular cortex subregional vulnerability to alpha-synuclein pathology in Parkinson's disease and dementia with Lewy bodies. Neuropathol Appl Neurobiol.
-
Halliday GM, Leverenz JB, Schneider JS, Adler CH (2014) The neurobiological basis of cognitive impairment in Parkinson's disease. Mov Disord 29: 634-650.
-
Jellinger KA (2012) Interaction between pathogenic proteins in neurodegenerative disorders. J Cell Mol Med 16: 1166-1183.
-
Siepel FJ, Bronnick KS, Booij J, Ravina BM, Lebedev AV, et al. (2014) Cognitive executive impairment and dopaminergic deficits in de novo Parkinson's disease. Mov Disord 29: 1802-1808.
-
Ekman U, Eriksson J, Forsgren L, Mo SJ, Riklund K, et al. (2012) Functional brain activity and presynaptic dopamine uptake in patients with Parkinson's disease and mild cognitive impairment: a cross-sectional study. Lancet Neurol 11: 679-687.
-
Christopher L, Duff-Canning S, Koshimori Y, Segura B, Boileau I, et al. (2015) Salience network and parahippocampal dopamine dysfunction in memory-impaired Parkinson disease. Ann Neurol 77: 269-280.
-
Huang C, Mattis P, Perrine K, Brown N, Dhawan V, et al. (2008) Metabolic abnormalities associated with mild cognitive impairment in Parkinson disease. Neurology 70: 1470-1477.
-
Bäckström D, Eriksson Domellof M, Granasen G, Linder J, Mayans S, et al. (2018) Polymorphisms in dopamine-associated genes and cognitive decline in Parkinson's disease. Acta Neurol Scand 137: 91-98.
-
Förstl H, Gratz S, Hahn U, Schwarz J, Jarnig M (2008) [Dementia with Lewy bodies and reduced dopamine transporter binding indicates significant acetylcholine deficiency]. Dtsch Med Wochenschr 133: S11-S14.
-
Bohnen NI, Albin RL, Muller ML, Petrou M, Kotagal V, et al. (2015) Frequency of cholinergic and caudate nucleus dopaminergic deficits across the predemented cognitive spectrum of Parkinson disease and evidence of interaction effects. JAMA Neurol 72: 194-200.
-
Liu AK, Chang RC, Pearce RK, Gentleman SM (2015) Nucleus basalis of Meynert revisited: anatomy, history and differential involvement in Alzheimer's and Parkinson's disease. Acta Neuropathol 129: 527-540.
-
Liu AKL, Lim EJ, Ahmed I, Chang RC, Pearce RKB, et al. (2018) Review: Revisiting the human cholinergic nucleus of the diagonal band of Broca. Neuropathol Appl Neurobiol.
-
Ray NJ, Bradburn S, Murgatroyd C, Toseeb U, Mir P, et al. (2018) In vivo cholinergic basal forebrain atrophy predicts cognitive decline in de novo Parkinson's disease. Brain 141: 165-176.
-
Schulz J, Pagano G, Fernandez Bonfante JA, Wilson H, Politis M (2018) Nucleus basalis of Meynert degeneration precedes and predicts cognitive impairment in Parkinson's disease. Brain 141: 1501-1516.
-
Hepp DH, Ruiter AM, Galis Y, Voorn P, Rozemuller AJ, et al. (2013) Pedunculopontine cholinergic cell loss in hallucinating Parkinson disease patients but not in dementia with Lewy bodies patients. J Neuropathol Exp Neurol 72: 1162-1170.
-
Woolf NJ, Jacobs RW, Butcher LL (1989) The pontomesencephalotegmental cholinergic system does not degenerate in Alzheimer's disease. Neurosci Lett 96: 277-282.
-
Kitajima Y, Hori K, Konishi K, Tani M, Tomioka H, et al. (2015) A review of the role of anticholinergic activity in Lewy body disease and delirium. Neurodegener Dis 15: 162-167.
-
Alexandris A, Liu AK, Chang RC, Pearce RK, Gentleman SM (2015) Differential expression of galanin in the cholinergic basal forebrain of patients with Lewy body disorders. Acta Neuropathol Commun 3: 77.
-
Albin RL, Dauer WT (2012) Parkinson syndrome. Heterogeneity of etiology; heterogeneity of pathogenesis? Neurology 79: 202-203.
-
Francis PT (2009) Biochemical and pathological correlates of cognitive and behavioural change in DLB/PDD. J Neurol 256: 280-285.
-
Del Tredici K, Braak H (2013) Dysfunction of the locus coeruleus-norepinephrine system and related circuitry in Parkinson's disease-related dementia. J Neurol Neurosurg Psychiatry 84: 774-783.
-
Jellinger KA (2007) Lewy body disorders. In: Youdim MBH, Riederer P, Mandel SA, Battistin L, Lajtha A, Degenerative Diseases of the Nervous System. Springer Science New York, 267-343.
-
Kalaitzakis ME, Pearce RK, Gentleman SM (2009) Clinical correlates of pathology in the claustrum in Parkinson's disease and dementia with Lewy bodies. Neurosci Lett 461: 12-15.
-
Espay AJ, LeWitt PA, Kaufmann H (2014) Norepinephrine deficiency in Parkinson's disease: the case for noradrenergic enhancement. Mov Disord 29: 1710-1719.
-
Vermeiren Y, De Deyn PP (2017) Targeting the norepinephrinergic system in Parkinson's disease and related disorders: The locus coeruleus story. Neurochem Int 102: 22-32.
-
Maillet A, Krack P, Lhommee E, Metereau E, Klinger H, et al. (2016) The prominent role of serotonergic degeneration in apathy, anxiety and depression in de novo Parkinson's disease. Brain 139: 2486-2502.
-
Ohno Y, Shimizu S, Tokudome K, Kunisawa N, Sasa M (2015) New insight into the therapeutic role of the serotonergic system in Parkinson's disease. Prog Neurobiol 134: 104-121.
-
Barker RA, Williams-Gray CH (2016) Review: The spectrum of clinical features seen with alpha synuclein pathology. Neuropathol Appl Neurobiol 42: 6-19.
-
Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, et al. (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24: 197-211.
-
Braak H, Bohl JR, Muller CM, Rub U, de Vos RA, et al. (2006) Stanley Fahn Lecture 2005: The staging procedure for the inclusion body pathology associated with sporadic Parkinson's disease reconsidered. Mov Disord 21: 2042-2051.
-
McKeith I (2009) Commentary: DLB and PDD: the same or different? Is there a debate? Int Psychogeriatr 21: 220-224.
-
Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, et al. (2009) Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol 117: 613-634.
-
Zaccai J, Brayne C, McKeith I, Matthews F, Ince PG (2008) Patterns and stages of alpha-synucleinopathy: Relevance in a population-based cohort. Neurology 70: 1042-1048.
-
Braak H, Del Tredici K (2008) Nervous system pathology in sporadic Parkinson disease. Neurology 70: 1916-1925.
-
Alafuzoff I, Ince PG, Arzberger T, Al-Sarraj S, Bell J, et al. (2009) Staging/typing of Lewy body related alpha-synuclein pathology: a study of the BrainNet Europe Consortium. Acta Neuropathol 117: 635-652.
-
Braak H, Del Tredici K (2017) Neuropathological staging of brain pathology in sporadic Parkinson's disease: Separating the wheat from the chaff. J Parkinsons Dis 7: 73-87.
-
Jellinger KA (2014) Neuropathology of Parkinson's disease. In: Thomas M (ed) Inflammation in Parkinson's Disease: Scientific and Clinical Aspects. Springer New York, 25-47.
-
Ferman TJ, Boeve BF, Smith GE, Lin SC, Silber MH, et al. (2011) Inclusion of RBD improves the diagnostic classification of dementia with Lewy bodies. Neurology 77: 875-882.
-
Brundin P, Melki R (2017) Prying into the prion hypothesis for Parkinson's disease. J Neurosci 37: 9808-9818.
-
Goedert M, Masuda-Suzukake M, Falcon B (2017) Like prions: the propagation of aggregated tau and alpha-synuclein in neurodegeneration. Brain 140: 266-278.
-
Longhena F, Faustini G, Missale C, Pizzi M, Spano P, et al. (2017) The contribution of alpha-synuclein spreading to Parkinson's disease synaptopathy. Neural Plast 2017: 5012129.
-
Goedert M, Eisenberg DS, Crowther RA (2017) Propagation of tau aggregates and neurodegeneration. Annu Rev Neurosci 40: 189-210.
-
Kovacs GG, Wagner U, Dumont B, Pikkarainen M, Osman AA, et al. (2012) An antibody with high reactivity for disease-associated alpha-synuclein reveals extensive brain pathology. Acta Neuropathol 124: 37-50.
-
Walker LC, Jucker M (2015) Neurodegenerative diseases: expanding the prion concept. Annu Rev Neurosci 38: 87-103.
-
Peng C, Gathagan RJ, Covell DJ, Medellin C, Stieber A, et al. (2018) Cellular milieu imparts distinct pathological alpha-synuclein strains in alpha-synucleinopathies. Nature 557: 558-563.
-
Melki R (2018) Alpha-synuclein and the prion hypothesis in Parkinson's disease. Rev Neurol (Paris).
-
Peelaerts W, Bousset L, Baekelandt V, Melki R (2018) a-Synuclein strains and seeding in Parkinson's disease, incidental Lewy body disease, dementia with Lewy bodies and multiple system atrophy: similarities and differences. Cell Tissue Res 373: 195-212.
-
Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, et al. (2015) alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522: 340-344.
-
Ouf A, Szigeti K (2017) Evidence-based review of therapeutic approaches in dementia with Lewy bodies. J Alzheimers Dis Parkinsonism 7: 1000406.
-
Palermo G, Ceravolo R, Bonuccelli U (2018) Advances in the pharmacotherapeutic management of dementia with Lewy bodies. Expert Opin Pharmacother: 1-11.
-
Rolinski M, Fox C, Maidment I, McShane R (2012) Cholinesterase inhibitors for dementia with Lewy bodies, Parkinson's disease dementia and cognitive impairment in Parkinson's disease. Cochrane Database Syst Rev.
-
Connolly BS, Fox SH (2012) Drug treatments for the neuropsychiatric complications of Parkinson's disease. Expert Rev Neurother 12: 1439-1449.
-
Boeve BF, Dickson DW, Duda JE, Ferman TJ, Galasko DR, et al. (2016) Arguing against the proposed definition changes of PD. Mov Disord 31: 1619-1622.
-
Gratwicke J, Zrinzo L, Kahan J, Peters A, Beigi M, et al. (2017) Bilateral deep brain stimulation of the nucleus basalis of Meynert for Parkinson disease dementia: a randomized clinical trial. JAMA Neurol.
-
Connors MH, Quinto L, McKeith I, Brodaty H, Allan L, et al. (2018) Non-pharmacological interventions for Lewy body dementia: a systematic review. Psychol Med 48: 1749-1758.
-
Schenk DB, Koller M, Ness DK, Griffith SG, Grundman M, et al. (2017) First-in-human assessment of PRX002, an anti-alpha-synuclein monoclonal antibody, in healthy volunteers. Mov Disord 32: 211-218.
-
Valera E, Masliah E (2016) Combination therapies: The next logical Step for the treatment of synucleinopathies? Mov Disord 31: 225-234.
-
Aarsland D, Creese B, Politis M, Chaudhuri KR, Ffytche DH, et al. (2017) Cognitive decline in Parkinson disease. Nat Rev Neurol 13: 217-231.
-
Brundin P, Dave KD, Kordower JH (2017) Therapeutic approaches to target alpha-synuclein pathology. Exp Neurol 298: 225-235.
-
Spencer B, Valera E, Rockenstein E, Overk C, Mante M, et al. (2017) Anti-alpha-synuclein immunotherapy reduces alpha-synuclein propagation in the axon and degeneration in a combined viral vector and transgenic model of synucleinopathy. Acta Neuropathol Commun 5: 7.
-
Postuma RB, Berg D (2017) The new diagnostic criteria for Parkinson's disease. Int Rev Neurobiol 132: 55-78.
-
Goetz CG, Emre M, Dubois B (2008) Parkinson's disease dementia: definitions, guidelines, and research perspectives in diagnosis. Ann Neurol 64: S81-S92.
-
Rizzo G, Copetti M, Arcuti S, Martino D, Fontana A, et al. (2016) Accuracy of clinical diagnosis of Parkinson disease: A systematic review and meta-analysis. Neurology 86: 566-576.
-
Rizzo G, Arcuti S, Copetti M, Alessandria M, Savica R, et al. (2018) Accuracy of clinical diagnosis of dementia with Lewy bodies: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 89: 358-366.
-
Joutsa J, Gardberg M, Roytta M, Kaasinen V (2014) Diagnostic accuracy of parkinsonism syndromes by general neurologists. Parkinsonism Relat Disord 20: 840-844.
-
Mouton A, Blanc F, Gros A, Manera V, Fabre R, et al. (2018) Sex ratio in dementia with Lewy bodies balanced between Alzheimer's disease and Parkinson's disease dementia: a cross-sectional study. Alzheimers Res Ther 10: 92.
-
Joki H, Higashiyama Y, Nakae Y, Kugimoto C, Doi H, et al. (2018) White matter hyperintensities on MRI in dementia with Lewy bodies, Parkinson�s disease with dementia, and Alzheimer's disease. J Neurol Sci 385: 99-104.
-
Cersosimo MG (2018) Propagation of alpha-synuclein pathology from the olfactory bulb: possible role in the pathogenesis of dementia with Lewy bodies. Cell Tissue Res 373: 233-243.
-
Irwin DJ, Hurtig HI (2018) The contribution of tau, amyloid-beta and alpha-synuclein pathology to dementia in Lewy body disorders. J Alzheimers Dis Parkinsonism 8: 1000444.
Citation
Jellinger KA (2018) Dementia with Lewy Bodies and Parkinson's Disease-Dementia: Current Perspectives. Int J Neurol Neurother 5:076. doi.org/10.23937/2378-3001/1410076