Leishmaniasis is a disease caused by the protozoan Leishmania that is considered one of the causes of death from parasitic infection worldwide. Looking for the right chemotherapy against leishmaniasis has been difficult because of the high toxicity of the most effective drugs. This disease is considered among the 13 unattended diseases worldwide according to the World Health Organization (WHO). In the present work, the leishmanicidal activity against L. amazonensis promastigote and cytotoxicity of 9 aryl-substituted imidazoles were evaluated. In vitro antileishmanial results indicated that the ligand 1, 2 and 7 exhibited strong activity against L. amazonensis promastigote, but only the compound 1 and 4 showed agood selectivity and cytotoxicity against the parasite. According to the results of the molecular docking, the aromatic substituents have mainly stabilizing hydrophobic interactions with the enzymatic matrix, which shows the probability that the protein trypanothione reductase is the therapeutic target of these compounds as possible antileishmanial drugs.
Aryl-substituted imidazoles, Leishmania amazonensis, Antileishmanial activity, Molecular docking
Leishmaniasis is a neglected disease caused by an intracellular protozoon transmitted by the bite of insects of the Phlebotominae subfamily (sandflies). Over 20 Leishmania species are known to be infective to humans. There are three main types of leishmaniasis: the most common clinical form is the cutaneous leishmaniasis (CL), whereas, the visceral leishmaniasis (VL) is more severe and often fatal if not treated. Moreover, the mucosal/mucocutaneous (MCL) has a chronic progression which may lead to deformities and long-term effects.This disease is among the endemics considered a priority in the world, being cited in 98 countries, affecting around 2 million people per year, with 350 million people being exposed to infection [1].
Only few drugs are available for the treatment of different forms of leishmaniasis. Pentavalent antimonials are of high cost and have severe side-effects [2]. These compounds are a first-line drug treatment for leishmaniasis, including sodium stibogluconate and meglumine antimoniate. Drugs, such as miltefosine, liposomal amphotericin B, pentamidine and paromomycin, have also been used to treat this disease [3]. Due to the problems associated with the severe adverse effects, high cost, drug resistance and the limited progress in developing a vaccine against human leishmaniasis, development of new drugs is highly required.
Nitrogen-containing heterocycles are ubiquitous in numerous biomolecules. The imidazoles are a vital class of compounds due to their prevalence in several biomolecules such as histidine, purines and natural products (alkaloids) [4,5]. These are considered as beneficial structural motifs by its wide range of applications, specifically those highly substituted imidazoles exhibit potent biological properties like antibacterial [6], anti-inflammatory [7,8], anticancer activities and work as neurogenic agents [9,10]. Drugs like fenflumizole®, losarton® and eprosartan® are some of the marketed drugs as substituted Imidazoles [9,10].
According to Rossi, et al. [11] several substituted imidazole with antileishmanial activity have been found. One tetrasubstituted imidazole has been reported as potential antileishmanial agent described by Prajapati and coworkers in 2015 [12]. They used virtual screening, molecular docking, ADMET, QikProp, MM-GBSA and molecular dynamics of 5-nitroimidazole analogues and concluded that the ethyl 2-acetyl-5-[4-butyl-2-(3-hydroxypentyl)-5-nitro-1H-imidazol-1-yl]pent-2-enoate has the great potential to develop an antileishmanial drug to kill L. donovani parasites. Other substituted imidazoles reported by Srinivas, et al. [13] in 2009 with antileishmanial activity against L. donovani. In addition, other recent studies reported by Martin, et al. [14] demonstrated the potential of imidazole salts as antileishmanial agents against promastigotes of L. amazonensis.
In the present study, we describe the leishmanicidal activity of different aryl-substituted imidazoles against L. amazonensis promastigote, as well as the application of the molecular docking technique, in order to explore the possible binding mode of aryl-substituted imidazoles to different targets from L. amazonensis. The compounds have already been reported to exhibit prominent antileishmanial activity against amastigotes of L. infantum (causal agent of VL) [15], but have not previously been tested against CL species such as L. amazonensis.
The chemical structures of nine aryl-substituted imidazoles are shown in Figure 1. The aryl-substituted imidazoles were obtainedby reaction ofaromatic aldehydes, primary amines, benzils, and ammonium acetate reported by our group in previous work [15]. The compound 1-benzyl-2-(3-nitrophenyl)-4,5-diphenyl-1H-imidazole (3), was synthesized following the same methodology reported in our previous article [15] and was confirmed through its physical and spectral properties.Melting points were determined in open capillaries using a Digital Melting Point apparatus WRS-2A (Jiangsu Zhengji Instruments Co., Ltd, China). 1H-NMR and 13C-NMR spectra were recorded with a Bruker DRX-500 spectrometer (Bruker, United Kingdom) obtained in DMSO-d6. The FT-IR spectra were recorded on a FTIR Infrared spectrometer (Shimadzu Corporation, Japan) in KBr (νmax in cm−1).
 Figure 1: Chemical structures of aryl-substituted imidazole.
View Figure 1
Figure 1: Chemical structures of aryl-substituted imidazole.
View Figure 1
Spectral data results for compound 3: 1-benzyl-2-(3-nitrophenyl)-4,5-diphenyl-1H-imidazole.MW: 431.16 mp: 150-152℃ (Reported: 151-153℃) [16]. FT-IR (KBr, cm−1): νmax3056(C = Carom), 1685(C = N), 1524(N-O), 1457 y 1500(C = Carom), 1340 (C-NO2). δmax 853(C-NAr-NO2), 710 y 810(C-Hm-sust); 1H-NMR (600 MHz, DMSO-d6): δ ppm 5.13 (s, 2H, CH2), 6.78-8.12 (m, 19H, Ar-H); 13C-NMR (150 MHz, DMSO-d6): δ ppm 51.88 (CH2), 122.0, 123.9, 127.43, 128.04, 128.29, 129.07, 129.20, 133.64, 134.3, 135.36, 136.93, 137.3, 144.4, 147,8, 148.4, 150.9, 153.7 (Carom); Mass spectrum (ES+): M (C28H21N3O2) calculated: 431.50 experimental: 431.6 [M + H]+, 386.15, 309.14, 219.9.
The nine compounds were dissolved in dimethylsulfoxide (DMSO, BDH, Poole, England) at a concentration of 20 ± 0.1 mg/mL in the stocks solutions for the biological studies.
Microorganism: Parasite strains were: L. amazonensis MHOM/BR/77/LTB0016 maintained in the culture collection of the Institute of Tropical Medicine Pedro Kourí, Havana, Cuba.
Antipromastigotes assay: In the first well of a 96-wells microplate with conical bottom (Greiner Bio-one, Germany), 2 µL of compounds extract was added to 98 µL of Schneider's medium (SIGMA, St. Louis, MO, USA) supplemented with 10% heat-inactivated fetal bovine serum (SIGMA) and penicillin-streptomycin as antibiotics (SIGMA); while 50 µL of medium were distributed in the other wells. Then, serial dilutions 1:2 were carried out to obtain final concentrations between 6.25 and 200 µg/mL. Afterwards, 50 µL of promastigotes were added at 4 × 105 parasite/mL in logarithmic phase. Microplates were sealed with parafilm and incubated at 26℃ for 72 hours. After incubation, 20 µL of a solution of 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT; SIGMA) at 5 mg/mL was added in each well. An additional incubation of 4 hours was performed, the supernatant was eliminated, and the formazan crystals were dissolved with 100 μL of DMSO. Finally, the microplate was read in an ELISA microplate reader (Sirio S Reader, 2.4-0, Italy) at 540 nm and 620 nm as reference wavelength [17]. The median inhibitory concentration (IC50) was determined by dose-response linear regression analysis. Each experiment was performed in duplicate and the results were expressed as the mean ± standard deviation.
Peritoneal macrophages: Resident macrophages from the peritoneal cavity were collected and washed with cold RPMI 1640 medium (SIGMA), supplemented with antibiotics (penicillin 200 IU, streptomycin 200 µg/mL). Then, 1-3 × 106 macrophages/mL were seeded in 96-well plates with conical bottom (Greiner Bio-one, Germany) and incubated for 2 hours at 37℃ and 5% CO2 atmosphere. The non-adherent cells were removed by washing with phosphate buffer solution. Subsequently, RPMI medium and compounds at the tested concentrations (6.25 -200 µg/mL) were added per well. The plates were incubated for 72 hours under the same conditions and macrophages treated with DMSO were included as control. The cellular viability was determined by colorimetric assay with 15 µL MTT, solution added per well. The median cytotoxic concentration (CC50) was determined by dose-response linear regression analysis. Each experiment was performed in duplicated and the results were expressed as the mean ± standard deviation.
The selectivity index (SI) was calculated using the IC50 (for protozoan) and CC50 (for macrophages) values obtained from each in-vitro assay [18], determined by the following Eq (1):
where CC50 is the 50% cytotoxicity concentration. Potential products are those that show an IC50 < 10 µM [19] and a SI > 5 [20] according to international criterions.
The molecular modelling was performed using the high-performance computing capabilities of the cluster of the Universidad de Oriente, Cuba (HPC-UO) (https://portal.uo.hpc.cu/website/). Molecular docking is one of the main methods of structure-based drug design (SBDD) molecular modeling, providing significant contributions in the discovery and optimization phases of leading compounds. Molecular docking studies were implemented in order to compare the interaction of the aryl-substituted imidazoles at the molecular level with different enzymes reported for the specie L. amazonenzis.
Ligands preparation: The three-dimensional geometries of the aryl-substituted imidazoles were drawn in ChemDraw Professional v.17.1.0.105 [21]. The structures were imported to Chem3D v.17.1.0.105 [21] and subjected to molecular energy minimization using the AM1 semiempirical method included in Chem3D.
Proteins preparation: The docking process was performed with the crystallographic protein structures downloaded from the RCSB Protein Data Bank (PDB). For the protein Arginase-I (ArgI), we used a previously described model of L. amazonensis arginase based on rat liver arginase complexed with AOH ((S)-2-amino-7oxoheptanoic acid) (PDB, ID code 1T5F) [22,23]. The enzyme trypanothione reductase (TryR) from L. infantum (PDB ID: 2JK6), TryR is a flavoprotein disulfide reductase dependent on NAPDH, and is found only in parasites of the Trypanosomatidae family. It is present in both L. infantum and L. amazonensis according to reference [24]. The nucleoside diphosphate kinase b (NDKb) from L. major (PDB ID: 3NGU) is a ubiquitous and conserved enzyme that plays a central role in the maintenance of intracellular nucleoside triphosphate (NTP) levels, and is also involved in the regulation of gene expression in mammalian cells, bacterial pathogenesis and parasite housekeeping [25]. These particular characteristics have made NDKs promising targets for the design and discovery of antiparasitic drugs, including antileishmanial agents.
The program AutoDock (v.4.2) was employed to perform docking experiments [26], keeping the protein fixed and allowing freely rotatable bonds for the ligands. The grid box was centered around the region of interest (active site) at the center-of mass of DHH ((S)-2-amino-7,7-dihydroxyheptanoic acid), ADP (adenosine-5'-diphosphate) and FAD (flavin-adenine dinucleotide) three-dimensional coordinates retrieved from PDB ID: 1T5F, 3NGU and 2JK6, respectively (Table 1). The Lamarckian Genetic Algorithm (LGA) implemented in the AutoDock program was applied using default parameters. The selection of docking ligand binding poses was based on the ranking of the fifty ligand conformational clusters and their respective binding energy values.
Table 1: Grid box parameters selected for the target enzymes. View Table 1
The binding site for proteins was identified from the analysis of the structures 1T5F, 2JK6 and 3NGU available in the RCSB Protein Data Bank. The active site of Arginase I (PDB ID:1T5F) involves the amino acids Ser137, Glu186, Asn130, Asp183, Asp128, His126, Asp124, Asp232, Asp234, Glu277 and His101 [23]. In Trypanothione Reductase (PDB ID: 2JK6) the active site residues are the amino acids Lys60, Ile199, Tyr198, Cys57, Arg287, Thr51, Pro336, Thr335, Ser14, Asp327, Thr160, Ala46, Asp35, Arg290 and Leu294 [27]. The Nucleoside Diphosphate Kinase b (PDB ID: 3NGU) active site is formed by Lys11, Tyr51, Leu54, Phe59, Leu63, Tyr66, Arg87, Thr93, Arg104, Val111, Gly112, Asn114, Gly118 and Asp120 [28].
To check the docking procedure and given the similar structure (imidazole and triazole rings) between the compounds here reported, we choose three known inhibitors reported. Compound ZINC12151998 (5-cyclopropyl-1- (6,7-dihydro-5H-benzo [6,7] cyclohepta [1,2-d] pyrimidin-2-yl) -N- (quinolin-6-ylmethyl) - 1H-pyrazole-4-carboxamide) proposed by Martinez, et al. [29] as a Trypanothione Reductase inhibitor, compound 2 - (5-methyl-2- (trifluoromethyl) - [1,2,4] triazolo [1,5-a] pyrimidin-7-yl) hydrazine-1-carbothioamide (compound 30) reported as Arginase I inhibitor by Da Silva, et al. [30] and compound (Z)-5-((5-chloro-2-oxoindolin-3-ylidene) methyl)-N-(2-(diethylamino)ethyl)-2,4-dimethyl-1H-pyrrole-3-carboxa-mide (SU11652) nucleoside diphosphatase kinase (NDK) inhibitor reported by Vieira, et al. [31], were rebuilt and redocked (Figure 2).
 Figure 2: Structure of control compounds A) ZINC12151998; B) Compound 30 and C) SU11652.
View Figure 2
Figure 2: Structure of control compounds A) ZINC12151998; B) Compound 30 and C) SU11652.
View Figure 2
Infections caused by parasites are today a serious problem in the health systems of many countries. Among these parasitic infections, leishmaniasis (caused by Leishmania spp.), Chagas disease (caused by T. cruzi) and African sleeping sickness (caused by T. brucei and T. rhodesiense) have high morbidity and mortality in tropical and subtropical regions of America, Africa and Asia, which are classified by WHO as neglected diseases. Nowadays, the search for natural products as an alternative treatment has motivated the interest of the scientific community.
In the present investigation the antiprotozoal activity for nine aryl-substituted imidazoles was evaluated against promastigotes of L. amazonensis, one of the species involved in diffuse cutaneous leishmaniasis in South America [32]. Our interest was based on its in vitro action against amastigote from L. infantum reported in our previous work [15]. The results are summarized in Table 2 and the calculated IC50 is reported, as well as the SI for the L. amazonensis model based on the CC50 obtained on the mouse peritoneal macrophages (host cells) determined directly in the experiment.
Table 2: Antileishmanial activity and cytotoxicity of aryl-substituted imidazoles. View Table 2
According to the analysis of the results, all compound tested are less active than amphotericin B. Compounds 1, 2 and 7 showed a marked activity against promastigotes of L. amazonensis with IC50 = 6.98 ± 0.8 μM, IC50 = 8.94 ± 1.0 μM and 6.72 ± 0.6 μM, respectively (Table 2). It is also observed in Table 2 that compounds 1 and 7 have a high selectivity index, demonstrating the selective over promastigotes.
Table 1 shows how the imidazoles 1, 2, 3 and 4, which have a similar structure (differ only in position 2 of the imidazole ring), have very different IC50 values. Compound 1 is the one with the best antileishmanial activity against L. amazonensis (IC50 = 6.98 ± 0.8 μM), while of the four the least active was compound 3 (IC50 = 16.22 ± 0.3 μM) and compounds 2 and 4 showed values of 8.94 ± 1.0 and 10.35 ± 1.2 μM, respectively. These results demonstrate how the influence of the phenyl-p-dimethylamino (compound 1) and phenyl-p-hydroxyl (compound 2) group in position 2 of the imidazolic ring increases the inhibitor power of the imidazoles.
The order of activity is 7, 1, 2 and 4, the first three meet the IC50 ≤ 10 μM criterion [33] to be considered as active compounds, although the last one is slightly higher. If Pink's criterion [34] is considered, these IC50 values should be between 2.3 and 2.4 μM. Therefore, it is recommended to continue its study due to the SI values and considering its toxicity. Taking these criteria into account, the imidazoles with the greatest potential to continue their studies are 1 and 4, since compound 7 is not favored by its greater activity due to its low selectivity index and its greater toxicity.
Compounds 1, 5 and 9 have in common the phenyl-p-dimethyl aminobenzaldeyde group as a substituent in the position 2 of the imidazole ring with very diverse activities, note how compound 9, which is a tri-aryl-imidazole, is inactive. The incorporation of an aryl substituent group in position 1 (tetra-aryl-imidazole) favors the activity, reaching the established activity criteria only for compound 1 with the benzimidazole substituent.
In the case of compounds 6 and 7, with structures much more similar to each other, they show a notable difference in activity. Both compounds possess the p-nitrophenyl substituent at the 2-position of the imidazole ring and differ in the position of the methyl group from the methylphenyl substituent at the 1-position of the imidazole ring (compound 6m-CH3 and compound 7p-CH3). Compound 7 is active while 6 is not active against L. amazonensis. However, in the previous study with L. infantum the result is inverse [15]. As shown in Table 2, there is a marked difference between the IC50 values determined against L. infantum and L. amazonensis, maintaining the same behavior only for compound 4 with similar IC50 values, so we can affirm that this behavior is due to the fact that the activities against both parasites were determined in different extracellular (promastigotes) and intracellular (amastigotes) forms.In addition, other recent studies reported by Martin, et al. [14] demonstrated the potential of imidazole in salts form as antileishmanial agents against promastigotes of L. amazonensis.
Molecular docking is a computational tool employed in the design and discovery of novel drug candidates, and was applied in order to understand the binding interactions of aryl-substituted imidazoles with the three selected targets: Arginase I, Trypanothione Reductase and Nucleoside diphosphate kinase b. The binding affinity of the docked molecules was evaluated by binding energy, calculated inhibition constants (Ki calc. by AutoDock 4.2), and hydrogen bonds in addition to the hydrophobic interactions with the enzyme pocket. The docked poses for each of the compounds have been evaluated and the pose with the lowest binding free energy and inhibition constant was thereby chosen (Table 3).
Table 3: In silico molecular docking results against the enzyme selected. View Table 3
Potential Trypanothione Reductase protein (TyrR) inhibitors: Based on our docking analysis, potential candidates that inhibit TryR are listed in the first column of Table 3, the binding energy (ΔG) and inhibition constant (Ki) of the control inhibitor Std-TryR were found to be -11.30 kcal/mol and 5.19 nM, respectively. However, the value for almost all aryl-substituted imidazoles is similar to Std-TryR, with ΔG values between -9.33 and -10.86 kcal/mol and Ki values in the order of nM. Compound 8 shows the best value for ΔG = -10.86 kcal/mol with a Ki = 11.00 nM and has a similar conformation as the control drug when binding to TryR (Figure 3). Other ligands as compound 3 and 4, showed also strong binding values of ΔG = -10.53 kcal/mol (Ki = 19.20nM) and -10.32 kcal/mol (Ki = 27.37 nM), respectively and those compouds have a similar binding conformation as compound 8 in the pocket of the enzyme TryR (Figure 4).
 Figure 3: Superimposition of the best pose of compound 8 (tan) + Std-TryR (sky blue) included in the binding pocket of Trypanothione reductase.
View Figure 3
Figure 3: Superimposition of the best pose of compound 8 (tan) + Std-TryR (sky blue) included in the binding pocket of Trypanothione reductase.
View Figure 3
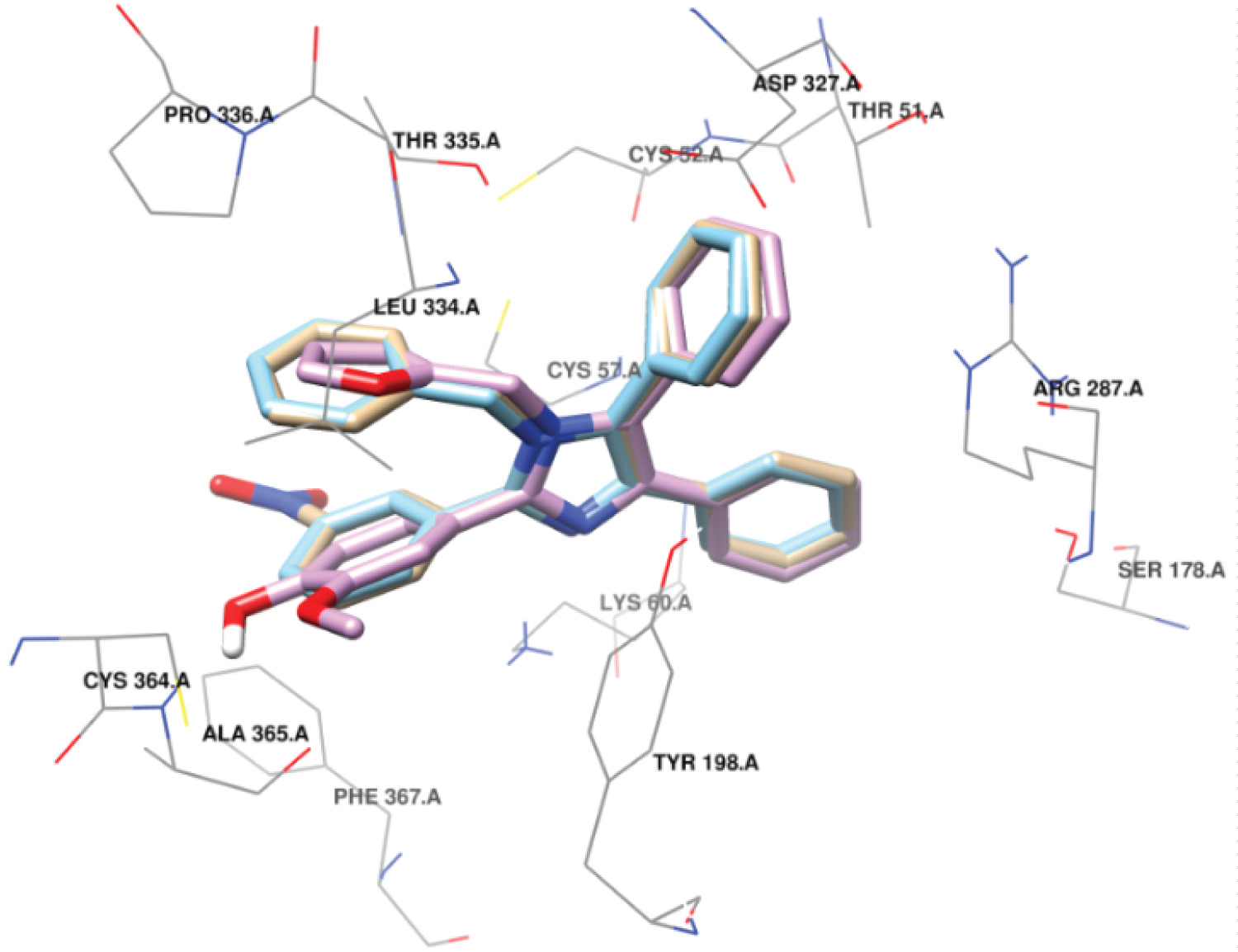 Figure 4: Superimposition of the best pose of compound 3 (tan) + compound 4 (sky blue) + compound 8 (orchid) included in the binding pocket of Trypanothione reductase.
View Figure 4
Figure 4: Superimposition of the best pose of compound 3 (tan) + compound 4 (sky blue) + compound 8 (orchid) included in the binding pocket of Trypanothione reductase.
View Figure 4
Three compound (3, 4 and 8) showed similar interaction in the binding pocket of TryR (Figure 5). Compound 3 was found to establish a hydrogen bond [N (pyridinic)....H-N] to the side chain of Lys60 with a distance of 2.88Å. The compound also shows hydrophobic interactions with Val55, Phe203, Gly56, Thr51, Leu334, Asp327, Leu334, Cys52, Pro336, Thr335, Tyr198, Cys57, Ala365 and Phe367 residues of this enzyme (Figure 5A). Compound 4 was bound in a hydrophobic pocket consisting of amino residues Ala365, Phe367, Cys57, Thr335, Pro336, Cys52, Asp327, Leu334, Thr51, Val55, Gly56 and Tyr198 and created one hydrogen bonding similar to compound 3 (with [N (pyridinic)....H-N] Lys60) (Figure 5C). Compound 8 interacted with Cys364, Leu334, Pro336, Thr335, Asp327, Thr51, Arg287, Cys52, Gly56, Val55, Phe203, Tyr198, Cys57, Phe367 by hydrophobic interaction. This compound establishes 2 hydrogen bonds with main chain NH and CO of Ala365 and [N (pyridinic)....H-N] to the side chain of Lys60 (Figure 5B).
 Figure 5: Docked structures of compounds A) 3, B) 8 and C) 4 in the Trypanothione reductase pocket represented using the LigPlot software [35].
View Figure 5
Figure 5: Docked structures of compounds A) 3, B) 8 and C) 4 in the Trypanothione reductase pocket represented using the LigPlot software [35].
View Figure 5
Potential Arginase I protein (ArgI) inhibitors: For the enzyme ArgI with the control drug Std-ArgI the binding energy was found to be -6.32 kcal/mol with a Ki= 23.38 μM. The values of Std-ArgI are exceded by all compounds (Table 3). The best ligand was 3 with ΔG = -10.28 kcal/mol and Ki = 29.14 nM being better than Std-ArgI. Also, the compounds 2 and 4 showed excellent values of ΔG with -9.55 and -9.33 kcal/mol and an inhibition constant of 99.7 and 143.84 nM, respectively higher than Std-ArgI.
Compound 3 showed hydrogen bonds [N(pyridinic)....H-O] with Thr246 and [N(amine)....H-O] with Arg21 residue of ArgI (Figure 6A) and hydrophobic interactions with Gly245, Asp232, Asp234, Ala141, Glu277, Asp128, His101, Asp124, Gly142; His126, Glu186, His126, Thr135, Asp183, Ser136, Asn130, Asn139 and Ser137. Some of those residues Asp232, Glu277, Asp128, His101, Asp124, Glu186, His126, Asp183, Asn130 and Ser137 are involved in the active site according to reference [25]. We can see in Figure 6B the superimposition between compound 3 and Std-ArgI. They share some important interacting residues as Asn130, Asp183, Asp128 and His126. Also, we can observe that compound 3 improves the interaction with the enzyme compared to Std-ArgI.
Potential Nucleoside diphosphate kinase b protein (NDKb) inhibitors: Nucleoside diphosphate kinases (NDKb) catalyze the transfer of a γ-phosphoryl group from a nucleoside triphosphate (NTP) donor to a nucleoside diphosphate (NDP) acceptor in a reversible mechanism. Thus, NDKb are considered to participate in different metabolic pathways, such as the regulation of gene expresión in mammalian cells, bacterial pathogenesis and parasite housekeeping. For this reason, the NDKb are promising targets for the design and discovery of antiparasitic drugs, including anti-leishmanial agents.
Potential inhibitors of NDKb are listed in the third column of Table 3, the binding energies (ΔG) and inhibition constant (Ki) for the control drug Std-NDKb were found to be -9.03 kcal/mol and 238.87 nM, respectively. However, all aryl-substituted imidazoles was found to be better binders than the reference compound Std-NDKb (except ligands 7 and 8), with ΔG values between -9.23 and -10.33 kcal/mol and Ki values in the nM range.
Compound 1 was to be found the best ligand by this molecular docking study, with ΔG = -10.33 kcal/mol and Ki = 26.82 nM, better than Std-NDKb. Compound 1 showed hydrophobic interactions with Tyr66, Leu63, Gly112, Ala62, Phe59, Val111, Thr93, Leu54, Arg104, Asn114, His117 and Gly118. Comparing those interactions with the reference, compound 1 and Std-NDKb share interactions with residues Tyr66, Leu63, Gly112, Phe59, Val111, Thr93, Leu54, Arg104 and Asn114 (residues whitin the red circles) involved in the active site [30-35] (Figure 7).
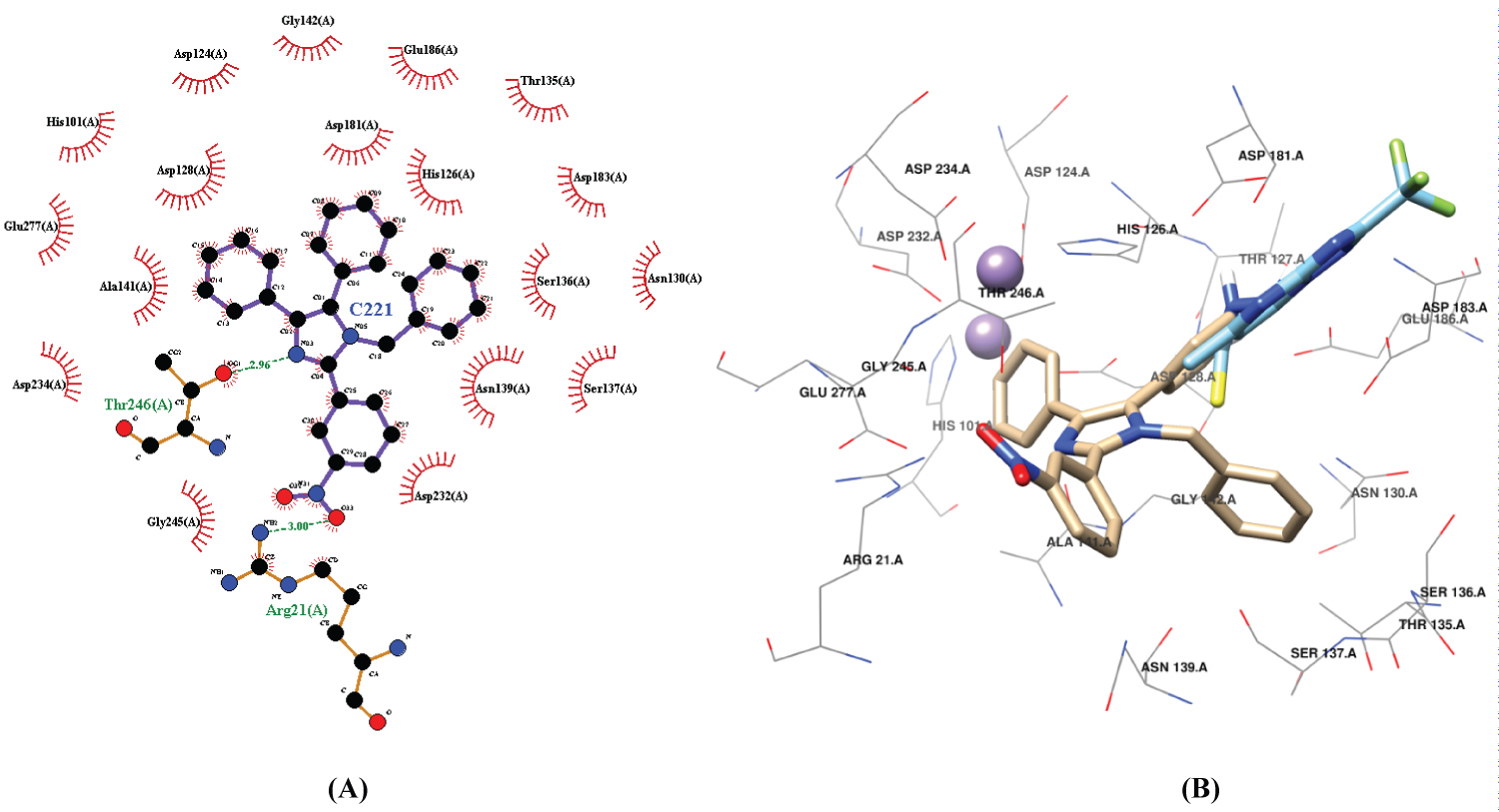 Figure 6: A) The interaction of compound 3 in the pocket of Arginase I using the LigPlot software [35]; B) Docked structures of compound 3 (tan) superimposed with Std-ArgI (sky blue).
View Figure 6
Figure 6: A) The interaction of compound 3 in the pocket of Arginase I using the LigPlot software [35]; B) Docked structures of compound 3 (tan) superimposed with Std-ArgI (sky blue).
View Figure 6
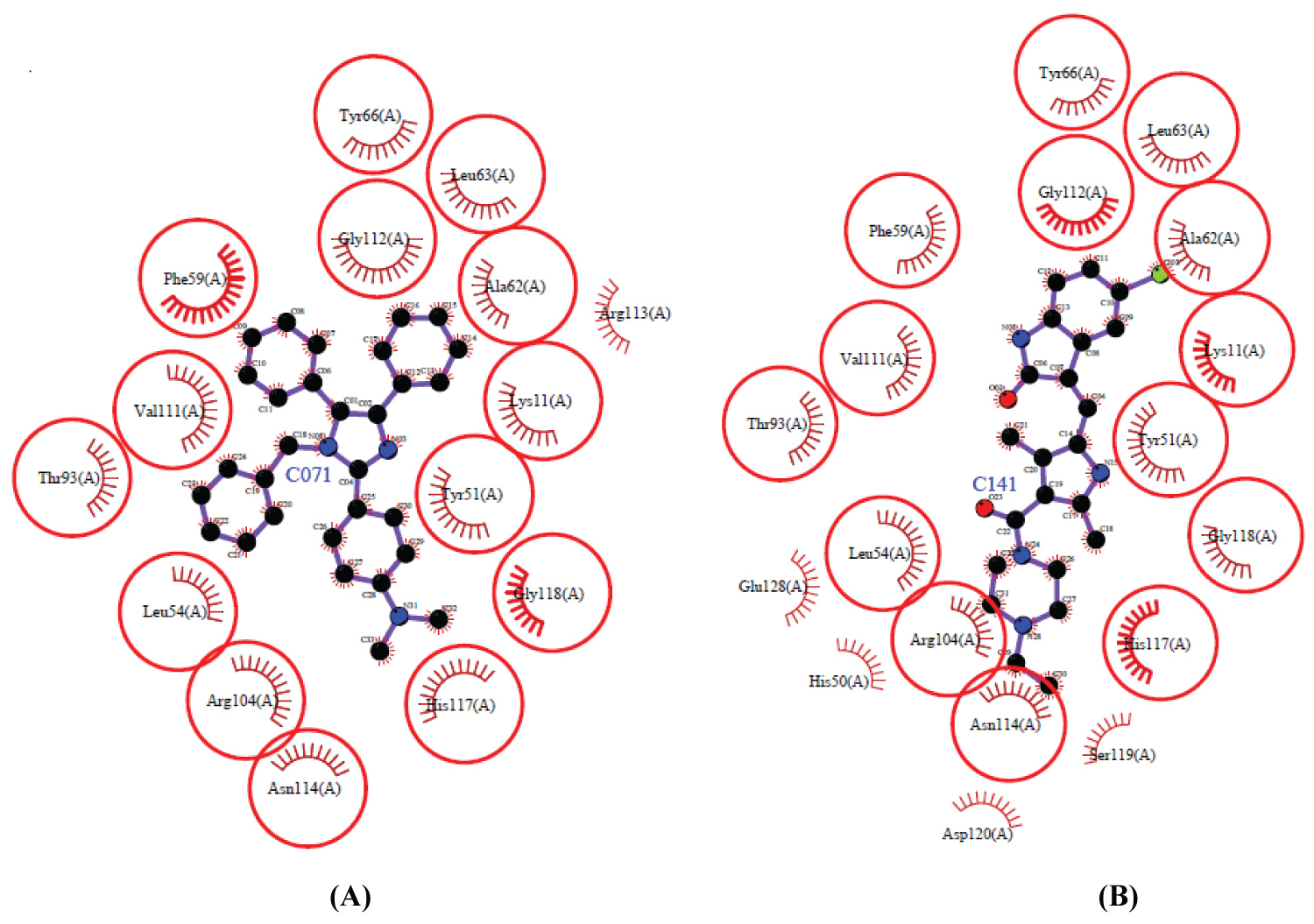 Figure 7: A) Interaction of compound 1 and B) Std-NDKb in the pocket of Nucleoside diphosphate kinase B) Represented using the LigPlot software [35].
View Figure 7
Figure 7: A) Interaction of compound 1 and B) Std-NDKb in the pocket of Nucleoside diphosphate kinase B) Represented using the LigPlot software [35].
View Figure 7
We identified the 4-(1-benzyl-4,5-diphenyl-1H-imidazol-2-yl)-N,N-dimethylaniline (1) and1-benzyl-2,4,5-triphenyl-1H-imidazole (4) as better antileishmanial activity against L. amazonensispromastigotes with the best selectivity index and low toxicity between the nine aryl-substituted imidazoles studied. The main interactions, studied by molecular docking, are hydrophobic between the substituents in positions 1, 4, and 5 of the imidazolic rings and residues in the active site. The analysis permitted us to show the potential responsible target for the antileishmanial activity to be the Trypanothione Reductase protein. The reported results can partly justify and support the possible use of these imidazoles for the treatment of the leishmaniasis caused by the protozoa L. amazonensis.