In 2015, we described short continuous fragments of human herpesvirus DNA, identical to the cellular ones, which we called microhomology (hits) because of their small size (≥ 20 nt). We noticed that generally the increase in the density (D) of these hits in human herpesviruses is inversely proportional to a decrease in the pathogenicity of these viruses. In this small work, we are considering the question of the existence of more objective features of HHV DNA (which can accompany the dynamics of the density of hits from HHV5 to HHV7), rather than a very imprecise notion of the degree of pathogenicity of a viral infection.
We show here that D really correlates with certain formal parameters of HHV DNA, primarily with their macrostructure, that is, with the number of segments of the HV genomes, their isomerization and size. According to these parameters, herpesvirus DNA can be divided into three groups, not strictly coinciding with the classification by subfamilies, genera and species. For a more detailed division (on species and strain level) according to the formal parameters discussed here and more convincing conclusions on the basis of the microhomology density between the viral and the host genomes, the number of completely sequenced HV DNAs is not enough for today. The situation is exacerbated by the low density of hits in the HV genome (5-20%) and their possible ambiguous functions. Some of the hits may have functional significance, as we suggested earlier [1,2], another part may be an accidental consequence of the close interaction of the viral and cellular genomes, for some reason fixed by selection. Nevertheless, the options for solving the task can be identified today.
Herpesviruses, Genome organization, Viral/cell DNA homology, Herpesviral episomes
Earlier we noticed that the DNA genomes of the viruses contain short (20-29 nt) continuous regions of nucleotide homology with cellular DNA, and that the maximum number of such sites occurs in natural host/virus pairs at the species level [2]. Because of the small size and scattered localization in the cellular genome, we called these sites microhomology, or hits. Later we showed that in the viral genome, the concentration of the hits correlates with the type of the virus and forms a sequence from HHV5 to HHV7 according to its increase [1]. We define the concentration of hits here as a percentage of the genome or gene occupied by hits, and we call this indicator the density of hits D. This approach avoids the difficulty in interpreting the long sections of viral DNAs formed by overlapping 20-member hits. The size of 20 nt is chosen by us because oligonucleotides of smaller sizes promptly increase the percentage of hits, and oligonucleotides of large sizes just as rapidly reduce them. In addition, this size approximately corresponds to the size of the siRNA, which may indicate the functional significance of the hits, which we wrote in 2010.
Characterization of herpesvirus pathology is very subjective, since all HHVs can cause both a general undefined symptomatology, associated primarily with the defeat of the functions of the immune system, and an acute generalized form of infection. However, according to a very rough estimate, the sequence of HHV5-HHV7 is characterized by a gradual decrease in the severity of HHV infection and its complications for people [1]. We then divided this sequence into three parts: I) Destructive pathology: HHV5,1,2,3; II) Proliferative pathology: HHV4,8, and III) Subclinical symptoms: HHV6,7; the third group consists of HHVs, whose direct involvement in a specific infectious pathology is yet to be revealed. Infections caused in humans by the viruses of the first group are cytomegalia, herpes (labial and genital) and chickenpox/zoster. The second group is infectious mononucleosis, Burkitt's lymphoma, nasopharyngeal carcinoma and Kaposi's sarcoma. The third group - roseoloviruses, causes ill-defined, symptomatology. It is only natural that the above-mentioned groups of infections were detected and investigated in the order of I-II-III, which is determined by the severity of the specific infection. A curious feature of this order is its compliance with the classification of HHV for subfamilies [3] with one exception: The polar positions of both genera beta-HB.
In this small work, we are considering the question of the existence of more objective features of HBV DNA (which can accompany the dynamics of the density of hits from HHV5 to HHV7), rather than a very uncomfortable notion of the degree of pathogenicity of a viral infection. Extremely insufficient information on the full genomic sequence of herpesviruses and their hosts, of course, reduces the credibility of this approach and makes any "horizontal" generalizations beyond the limits of HHV (or HV in general) nor the "vertical" evolutionary interpretation too reliable. We consider here the most general characteristics of HV genomes, without relating them to the molecular biology of replication and other viral syntheses.
Biological regularities are usually not mathematically strict, although at least one comprehensive morphological distinction of all types of order of Herpesvirales [4] from all other known viruses (except the tailed ones, which are considered their predecessors [5] is completely formal. This is a unique structure of the HV capsid, an icosahedron with a triangulation number of T = 16.
Herpesvirus DNAs are more diverse than the capsid, they can differ at a lower taxonomic level, that is, at the level of families, subfamilies, genera and even types (species). The most common characteristic of herpesvirus DNAs are their size and macrostructure, the elements of which are segments and terminal repeats. The macrostructure of the HV genome determines its class [3]. The species and strain differences between these viruses are more detailed and are determined by the nucleotide and amino acid sequences of the viral genes and their products, the GC content, etc.
According to macrostructure the HHV DNA is traditionally divided into 6 classes - from A to F [6]; outside the HHVs this classification can be somewhat extended (today GenBank contains almost 90 types of HV with fully sequenced genomes). On the basis of segmentation, HHVs can be divided into two large groups - one- and two-segment. Here we define the segment as a unique nucleotide sequence bounded by identical nucleotide sequences (repeats) having either a common (classes A, B and C, Figure 1) or mutually inverted (if it consists of two segments, each of which is bounded by a terminal and internal repeats, classes E and D) direction. Class F combines either original HV DNAs, or non-segmented variants (strains, isolates, etc.) that have a segmented prototype. Classes B and C differ from class A by the presence of a tandem series of terminal repeats, the non-strict number of which for class B (~35-45) and for class C (~4) differ by an order of magnitude. The cluster of internal genomic repetitions, which differ from terminal ones (shown in Figure 1 by a lesser height), leaves viral DNA of class C a single-segment one.
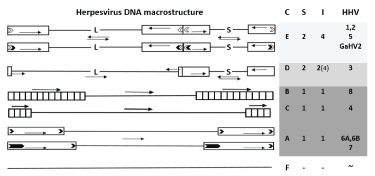 Figure 1: Basic macrostructural classes of HHV DNA.
Figure 1: Basic macrostructural classes of HHV DNA.
The length of the fragments of the genome is not met. The order of genomes with different macrostructure (from top to bottom) corresponds to the increase in the density of hits. Two-segment genomes with four segment inversions (class C) - Light-colored cells, with two inversions (class D) - Gray cells, single-segment genomes (classes A, B, C) - Dark gray cells, unsegmented genomes (class F) - colorless cells. Columns to the right: C - HHV classes by Pellett and Roizman; S - the number of segments; I - The number of isomers. Rectangles show terminal and internal inverted repeats; L - Long unique segment, S - Short one. Arrows show inversions of genomic regions. Light corners show sequences a', dark ones - telomere sequences. The large size of the telomere sequences of the SHG7 is emphasized by the greater extent of the dark corner.
View Figure 1
Directions of unique sequences of both segments of class E DNA relative to each other can be equal, thus forming four equimolar isomers. Short terminal repeats of the L segment of the D genomes do not allow such equimolarity to be observed; 95% of the population of HHV3 remain two-isomer due to free inversion of the short segment, and only 5% allow the inversion of the long segment. Single-segment genomes (classes A, B and C) form only one isomer.
Outside the HHV, the macrostructure character varies somewhat more, remaining mainly within the Pellett and Roizman classes. For example, if a long segment is bounded by a very short (less than 100 nucleotides) terminal/internal repeats in a two-segment VZV DNA, then in Bovine alpha herpesvirus 5 it can be completely deprived of them [7], remaining an L-segment variant based on a common set of genes with other representatives of the class D. In the DNA repeats of some HVs, telomere-like or other special sites are inserted, for example, a'. There are other HV of the Herpesviridae family and the Herpesvirales order, whose DNA structure, despite its unusual nature, can nevertheless be conventionally attributed to one of the described classes; we do not consider them here because we could not examine their genomes for the presence and density of hits.
The classification above (Figure 1) summarizes only those data that GenBank contains, and obviously has a current nature. Recall also that the composition of the subfamily does not necessarily include genomes of the same macrostructural subgroup. An example is CMV and roseoloviruses. We believe, however, that the genomic macrostructure of HV DNA is an evolutionary feature already at the genus level, and it should be taken into account when constructing phylogenetic trees [8-12] along with more detailed signs - the sequence and function of individual (orthogonal, primarily) genes or their complexes - because it refers to the whole genome, and the sequence and functions of cluster of orthologous group (COG) - to individual genes or to their relatively small groups [13,14].
It also makes sense to note that the segmentation of the genome can characterize only a part of the Herpesviral population of the same species. HHV5 strain Merlin NC_006273.2 belongs to class E, while, for example, strain NL/Rot6/Nasal/2012 [15] of the same virus is not segmented (class F). The number of repetitions can also be influenced by the number of passages in cell line after primary isolation (strains AD169 and Toledo of the CMV virus, GenBank). The number of tandem end-repeats of genomes HHV4 and HHV8 (classes B and C) is also not fixed, but always preserves a sharp difference between both types of viruses.
We analyzed the density of the hits in the DNA of each of the nine types of HHVs and summarized the results in Table 1. For each strain or isolate of these types, two to six variants of full-length DNA are analyzed. This not too large number corresponds to the maximum number (two) of fully sequenced genomes of HHV7. Table 1 also includes data on several types of HV of monkeys and birds (dark cells), which can be conditionally compared with the host genomes of monkeys and birds, whose full sequence is also contained in GenBank.
Table 1: HV DNA Analyzed in this paper. View Table 1
As an object of research, we analyzed this time not a viral genome in the narrow sense of the word (exome), that is, a sequence of viral genes (exons) occupying up to ~80% of the whole length of the HHV DNA, but DNA (including untranslated regions) in which these genes can overlap one another, be interrupted by introns, localized in complementary chains, or absent at all. This approach, as it turned out, slightly increases the density of hits in viral DNA. The computer algorithm for identifying hits can be found in open access repository Bitbucket.org [1].
Table 1 shows the density of HHV DNA hits, generally confirming the order that we showed in 2015. Recall that the assumption of the relationship between the density of hits and the degree of pathogenicity of the virus is incorrect already at the subfamily level, since in this case the most and least pathogenic viruses localize in common subfamily, whereas at the level of genus they occupy opposite positions (cytomegalo- and roseoloviruses). Mardivirus, which causes severe infection in its hosts (chickens), has the lowest density of hits, as does the virus B, which causes an even more severe pathology in monkeys, correspond to the alpha-HV subfamily.
We never found 20-nt hits in the mitochondrial DNA of the host. Our results are hardly accidental, as we showed earlier [2] comparing the microhomology of viral and human genomes. In this paper, we tried to understand whether the hits density D corresponds to the macrostructural characteristics of HV DNA, more or less free from subjective interpretation.
We believe that the interaction of complementary sites of HV DNA may be one of the factors determining the behavior of this molecule at the stages of its replication. In single-segment DNA, the presence of long unidirectional regions, especially those that are complementary to regions of host chromosomes (for example, telomeric ones), should apparently facilitate interaction with the host genome, from local homologous recombinations to full-format insertions. Two-segments DNAs can also contain unidirectional sites (either a', HHV1, or telomere-like, GaHV3) at both ends of the molecule. Complementarity, albeit incomplete, of these segments of the host's DNA should seemingly contribute to the insrtion of the complete viral genome in the host one, just as in single-segment HHV DNAs. Such an insertion does indeed occur (GAHV3, whose terminal and internal DNA repeats contain telomeric regions), but in the simplex viruses the situation is different: The complementarity of their TRs, including the a' regions, to the host genome is not very strict, and the HSV genome has not been inserted into the host genome.
We have analyzed terminal sections of HV lacked genes (such areas are denoted here by the symbol H). The results of the analysis are summarized in Table 2 and are partially illustrated in Figure 2. It turned out that these sites are accented (by high density of hits) almost in all types of HHV with a segmented genome, except for CMV. They are, first of all, responsible for increasing the density of hits in full-length HHV DNA.
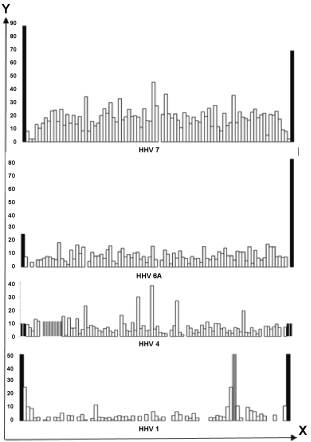 Figure 2: The density of hits (Y-axis) in the H-sections (Table 2) of some of the HHV genomes as compared to the same index for the genes (X-axis) of each of these HHVs.
Figure 2: The density of hits (Y-axis) in the H-sections (Table 2) of some of the HHV genomes as compared to the same index for the genes (X-axis) of each of these HHVs.
Black color - N-hits, gray - hits of internal inverted (HHV 1) or tandem (HHV 4) repetitions. Only vertical proportions of the size of HHV genome hits are observed.
View Figure 2
Table 2: Characteristics of terminal repeat sites of HV DNAs, lacked genes (H-SITES). View Table 2
Our data confirm the data published before (1) about the increase in the density of D' hits in the genomes (exomes) of herpesviruses from HHV-5 to HHV-7 and extends this regularity to the complete nucleotide sequence of the Herpesviral DNA. The density of hits D in total HV DNA slightly increases, in comparison with the D' density, but retains the previously shown series HHV5,1,2,3,8,4,6B,6A,7 somewhat disturbed by high indices for HSV1 and 2 (Figure 3).
The density of hits in Figure 3 demonstrates the obvious parallels with the macrostructure of HHV DNA. In the left part of the diagram (HHV5,1,2.3) HVs with two-segment genomes are located, in the right one (HHV8,4,6B,6A,7) - with single-segment genomes. The same applies to the HV genomes of monkeys and birds, which we were able to analyze. It seems that mutually complementary TP and IR of the two-segment genomes will, when replicating or forming an episomes, rather mate with each other [16] than interact with cellular DNA, which is characteristic of single-segment genomes with their direct terminal repeats. The direct terminal repeats, which seem to increase the probability of insertion of HHV6A and 6B into the host genome, are also characteristic of two-segment HV genomes (a' sites), however, the sizes of a' sites (250-600 nt) are much less than HHV6,7 TRs (8.000-10.000 nt); the a'-sites can be used to close linear DNA, for example, like TR in HHV4 [17]. The fact that such an insertion is not known in HHV7 (whose genome is organized similar to HHV6), is most probably explained either by insufficient informayion or by "looseness" of telomere regions in comparison with HHV6, where these sections are continuous for 120 - 200 nt.
In gamma-HHVs, the relatively small size of terminal repeats (500-800 nt) is compensated by their tandemization, and in EBV DNA it is "amplified" by additional intra-genomic tandem repeats (Figure 2). Such organization of the viral genome possibly promotes interaction with the cellular one to a greater extent than free of internal tandem repeats of comparable size of HHV4 DNA.
In the two-segment HHV3 genome the terminal hits have a density that is lower than in the HHV1 and 2, and the repeats-TR and IR lacked of a' regions are of short length, especially L-segment repeats (< 100 nt). The HHV3 S segment is even shorter than its own IR and TR repeats. Probably due to these features the HHV3 genome has mainly 2 isomers and differs from HHV1, 2 not having comparable ability to screen areas with the largest D hits from interaction with host DNA, which enhances the interchange with it of short fragments. For these reasons, the two-isomeric HHV3 genome occupies a position intermediate between one- and four-isomer genomes.
Based on the above observations, it can be concluded that the HHV full-genome insertion is not a necessary condition for increasing density of hits in the HV genome. Two facts suggest this.
1. The ability of the genome of GaHV2 to insert into the cell one by telomer-like sequences is not accompanied by an increased density of the hits, although in H-sites this density approaches the roseolovirus.
2. The insertion into human chromosomes is not shown for the genome of HHV7 with the highest density of hits. The high hits density in the herpesvirus macaque nemestrines is most likely not associated with its insertion into host DNA supported by telomeric sequences, which are even more "loose" than in HHV7; at the same time, the density of H-region hits in this virus is comparable to HHV7 (much higher than for HHV6).
The presented data show that the macrostructure of HHV DNA is in a certain correspondence with the density of genomic (exomic) hits in these viruses. Three components of the HV DNA macrostructure considered here are a) The overall size of the molecule; b) The number of its segments, and c) Flanking them repeats, which may contain specific regions - a' and H.
First and foremost, this correspondence is expressed as fact that the density of exomic hits in HHV with two-segment DNA is higher than that of HV with single-segmented ones. Randomness of such a "rule" requires further analysis. It is necessary, for example, to explore the hits density of the genomes of the CMV genome of Maastricht rats (beta-HV), which consists of one segment bounded by direct TRs (class A), although it has CMV-size DNA [18].
It can be assumed that in two-segment HHV DNA, a close interaction with the cellular genome, which strengthens with H sections, is significantly weakened by the mutual complementarity of repetitions flanking the segments. In HHV3 DNA, this effect is weaker, since L-segment terminal repeats are very short (< 100 nt). The proximity of the density of exomic hits in HHV3 and HHV8 (at the junction between one- and two-segment HBV DNA) places HHV3 in an intermediate position between both groups, which is emphasized by the number of genomic isomers (two), different from that in two-segment genomes (four) and single-segment (one).
It makes necessary to further investigate D separately for the genera of each HV subfamily and to find out that in alpha and beta-HHVs, the hits density (in general and in the H regions) increases with decreasing genome sizes (from simplexes to zoster viruses, and from cytomegalo- to roseoloviruses). In gamma-HHV the situation is different and depends, perhaps, on those considerations that we cited above. Incidentally, this parallel between the size of the genome and the density of hits is also noticeable in the separate analysis of both HHV groups - with two- and with single-segment genomes (including gamma-HHV). The further researches that partially extends out the limits of the HV order should help to exclude the probable eventuality of all these observations.