The apolipoprotein E4 (APOE4) allele represents the single greatest risk factor for late-onset Alzheimer's disease (AD) and accumulating evidence suggests that fragmentation with a toxic-gain of function may be a key molecular step associated with this risk. Recently, we demonstrated strong immunoreactivity of a 151 amino-terminal fragment of apoE4 (E4-fragment) within the nucleus of microglia in the human AD brain. In vitro, this fragment led to toxicity and activation of inflammatory processes in BV2 microglia cells. Additionally, a transcriptome analysis following exogenous treatment of BV2 microglia cells with this E4 fragment led to a > 2-fold up regulation of 1,608 genes, with many genes playing a role in inflammation and microglia activation. To extend these findings, we here report a similar transcriptome analysis in BV2 microglia cells following treatment with full-length ApoE4 (FL-ApoE4). The results indicated that full-length ApoE4 had a very small effect on gene expression compared to the fragment. Only 48 differentially expressed genes (DEGs) were identified (p < 0.05, and greater than 2-fold change). A gene ontology analysis of these DEGs indicated that they are not involved in inflammatory and activation processes, in contrast to the genes up regulated by the E4-fragment. In addition, genes that showed a negative fold-change upon FL-E4 treatment typically showed a strong positive fold-change upon treatment with the fragment (Pearson's r = -0.7). Taken together, these results support the hypothesis that a key step in the conversion of microglia to an activated phenotype is proteolytic cleavage of FL-ApoE4. Therefore, the neutralization of this amino-terminal fragment of ApoE4, specifically, may serve as an important therapeutic strategy in the treatment of AD.
Apolipoprotein E4, Fragment of ApoE4, Microglia cells, BV2 cells, Alzheimer's disease, Inflammation, Toxicity, RNA-seq, Transcriptome analysis
Harboring the apolipoprotein E4 (APOE4) allele represents the greatest genetic risk factor for late-onset Alzheimer's disease (AD) [1]. In this respect, carrying a single APOE4 allele increases AD risk 3-fold while those who are homozygous for the gene have a greater than 10-fold risk of developing AD [2]. The function of ApoE4 involves lipid transport within the CNS, although multiple other roles have been postulated including signaling through cell surface receptors, and modulating synaptic function [3]. However, to date, none of the classic roles of ApoE4 have satisfactorily explained how this protein increases dementia risk. One possible mechanism is a toxic-gain of function of amino-terminal domains of ApoE4 following cleavage by extracellular proteases [4].
We have recently identified a 151 amino-terminal fragment of ApoE4 (E4 fragment) that is generated following cleavage of the full-length ApoE4 (FL-ApoE4) and localizes within the nuclei of microglia cells in the human AD brain [5]. In addition, we have shown that exogenous treatment of BV2 microglia cells with this E4 fragment leads to the uptake and trafficking to the nucleus and cytotoxicity [5]. A transcriptome analyses following exogenous treatment with the E4 fragment resulted in the up regulation of almost 4,000 genes, with 20 of these genes up regulated 182- to 715-fold. The majorities of these genes have strong inflammatory properties and play a role in the polarization toward microglial M1 activation [6].
To extend these findings, we here investigated whether or not exogenous treatment of BV2 microglial cells with full-length apoE4 would lead to similar gene expression changes. Surprisingly, transcriptome analysis revealed little change in gene expression in cells treated with FL-ApoE4, relative to non-treated controls. In total, there were only 48 differentially expressed genes (DEGs, fold change > |2|, p < 0.05), many of which are known to carry out normal physiological roles. Importantly a comparison of differentially expressed genes in both studies showed that genes with a negative fold change upon FL-E4 treatment typically showed a strong positive fold change upon treatment with the fragment (Pearson's r = -0.7). These anticorrelated genes, which were up regulated by the E4 fragment, included several inflammatory mediators such as Il27, Il23a, and Gdf15. Collectively, these data indicate that fragmentation of FL-ApoE4 is a key determinant to the activation of inflammatory pathways and thus may serve as the potential link to the enhanced dementia risk associated with this gene.
BV2, murine microglial cells, were maintained at 37 °C and 6% CO2 in a humidified incubator. Cells were maintained in RPMI 1640 Media (Hyclone) supplemented with 10% standard fetal bovine serum (Hyclone), 10% Cellgro MEM Nonessential Amino Acid (Corning) and 10% Penicillin streptomycin (Hyclone). Cells were cultured in 50 mL T25 Flasks. All supplies were purchased from Thermo Fisher Scientific Inc. (Waltham, MA). For immunocytochemical studies, cells were plated in Falcon chamber cell culture slides for 24 hours prior to treatment. Treatment consisted of addition of either 25 μg/ml of full-length ApoE4 or nApoE41-151 for 24 hours in media. Control cells (untreated) had an equivalent level of conditioned media added to the wells.
For RNAseq experiments, treatment of BV2 cells was undertaken by incubation with the human FL-ApoE protein (Prosci Inc., Poway, CA) at a concentration of 25 μg/ml for 5 hours to assess changes in mRNA expression. Construction and purification of the amino-terminal fragment 1-151 for ApoE4 (nApoE41-151) was contracted out to GenScript (Piscataway, NJ). For this protein, a 6X-His tag was coupled to the fragment to facilitate purification.
Following treatment studies, BV2 cells were fixed by incubating cells in 4% paraformaldehyde for 23 minutes. For antibody labeling, cells were washed with 0.1M Tris-buffered saline (TBS), pH 7.4, and pretreated with 3% hydrogen peroxide in 10% methanol to block endogenous peroxidase activity. Slides were subsequently washed in TBS with 0.1% Triton X-100 (TBS-A) and then blocked for thirty minutes in TBS-A with 3% bovine serum albumin (TBS-B). Slides were further incubated overnight at room temperature with anti-His rabbit antibody (1:2,000) or an anti-ApoE4 N-terminal antibody (1:500, Aviva Biosciences). Following two washes with TBS-A and a wash in TBS-B, slides were incubated in anti-rabbit HRP-Conjugated secondary antibody. Visualization was accomplished by using a tyramide signal amplification kit (Molecular Probes, Eugene, OR) consisting of Alexa Fluor 488-labeled tyramide (green, Ex/Em = 495/519). Slides were mounted using Prolong Gold Antifade Mountant with DAPI (Molecular Probes).
Total RNA was extracted from cells with the Direct-zol RNA MicroPrep Kit (Zymo Research Corp., CA, USA) according to manufacturer's instructions. Genomic DNA was eliminated using TURBO DNAse as described by the manufacturer (Life Technologies, CA). RNA quality was assessed using spectrophotometry and gel electrophoresis. Total cDNA was generated from 1 μg of total RNA using qScript cDNA SuperMix (QuantaBio, MA, USA). Prior to use in qPCR, cDNA was diluted 1:2 with water.
RNA-sequencing was performed by the Molecular Research Core Facility at Idaho State University (Pocatello, ID). All samples were sequenced using an Illumina HiSeq4000 Sequencer. Library preparation was carried out using NEBNext Ultra II Directional RNA Library Prep Kit. Reads of 1 × 76 bp were assessed for quality using FASTQC v0.11.8. No sample reads were flagged for poor quality and all passed adapter content analysis indicating adapters were sufficiently removed from the reads during sequence processing. Consequently, no read trimming was applied to the samples. Using Hisat2 v2.2. 1 [7] a mouse reference genome (GRCm39 obtained from NCBI) was built and reads were mapped to this reference. Gene counts were determined using HTSeq v0.13.5 [8] (with parameters: '--type=gene --idattr=Name --stranded=no') after which, read counts were normalized using the median-of-ratios method implemented within Deseq2 v1.30.1 [9]. Deseq2 was then employed to calculate p-values using a Wald test with a Benjamini-Hochberg post hoc correction. Genes with an adjusted p-value < 0.05 and fold change ≥ |2| (log2 fold change ≥ 1) were considered to be differentially expressed. For down regulated genes, fold change was reported as -(2|log2 fold change|). The specific codes and implementation steps are available for review at https://gitlab.com/bsu/biocompute-public/apoe4_fl. The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus (GEO) with the identification number of GSE173381: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE173381.
A list of all DEGs and their respective fold changes was imputed into the PANTHER classification system [10,11]. This process was performed for biological processes and pathways [10], and the respective outputs were used. All presented data were statistically significant with a corrected p-value < 0.05.
As an initial approach, we examined the subcellular localization of ApoE4 in BV2 microglia cells following exogenous treatment. In this case, BV2 cells were treated with either a sublethal concentration of nApoE41-151 (25 μg/ml) and an equivalent concentration of full-length ApoE4 for 24 hours prior to immunocytochemical studies. As shown in Figure 1, strong nuclear localization of the nApoE41-151 was evident (Figure 1C), while localization of full-length ApoE4 appeared to be more cytoplasmic although some nuclear staining was also evident (Figure 1E). These results confirm our previous findings in vitro [5].
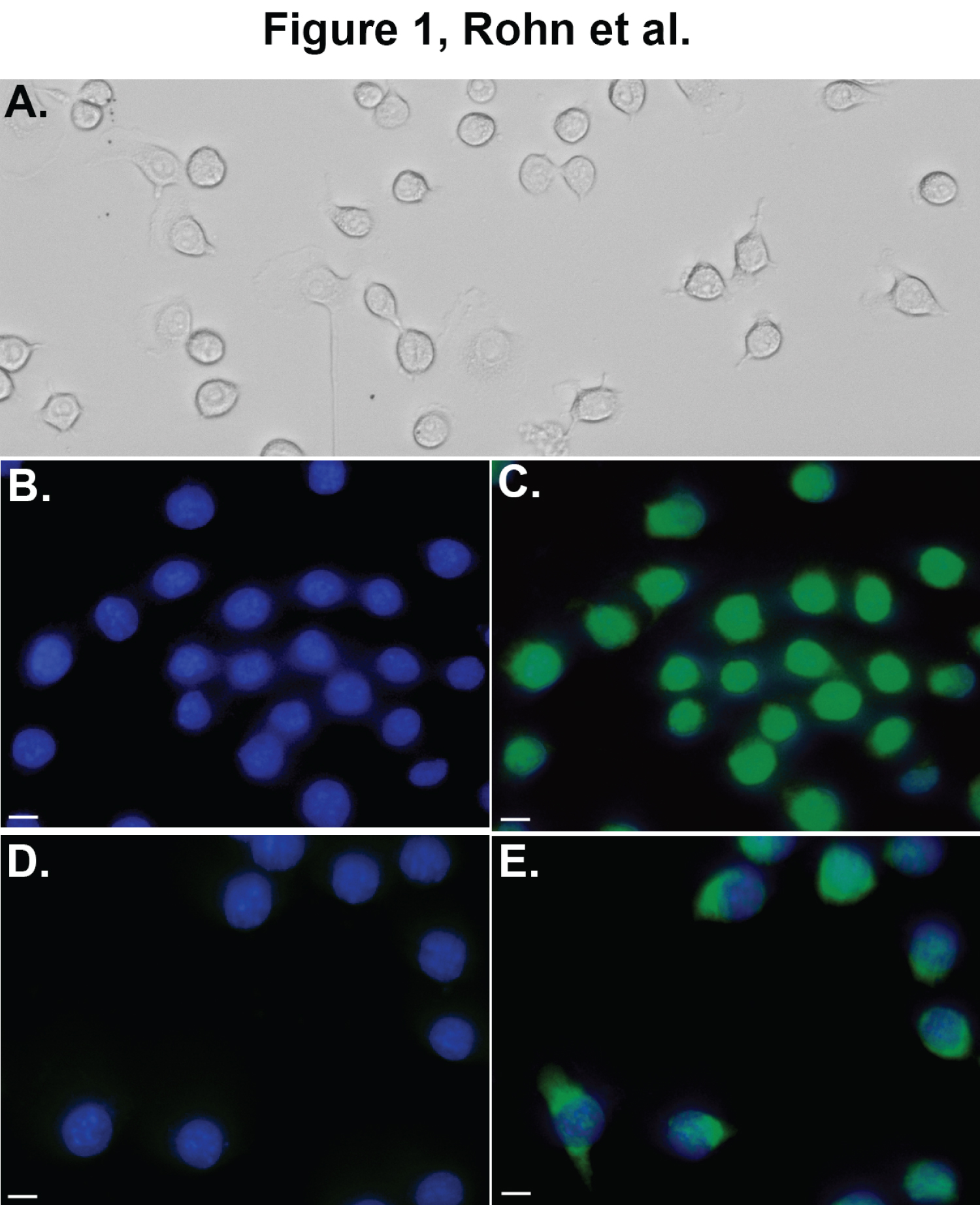 Figure 1: Nuclear localization of nApoE41-151 in BV2 microglia cells. Panel A is a representative phase-contrast, bright field image depicting characteristic morphology of healthy BV2 cells used in this study. Panels B-E depict representative double-label, fluorescence staining following treatment of BV2 cells with either 25 μg/ml nApoE41-151 (B and C, green) or full-length ApoE4 (D and E, green) for 24 hours. Nuclear staining with DAPI is indicated in blue with overlap images of both markers shown in either Panel C or E. Detection of 25 μg/ml nApoE41-151 was obtained using an anti-His antibody while detection of full-length ApoE4 was carried out using anti-ApoE4 N-terminal antibody. Strong nuclear localization of nApoE41-151 was evident (Panel C) following exogenous treatment, while the majority of labeling of full-length ApoE4 appeared to be cytoplasmic (Panel E). All scale bars represent 10 μm.
View Figure 1
Figure 1: Nuclear localization of nApoE41-151 in BV2 microglia cells. Panel A is a representative phase-contrast, bright field image depicting characteristic morphology of healthy BV2 cells used in this study. Panels B-E depict representative double-label, fluorescence staining following treatment of BV2 cells with either 25 μg/ml nApoE41-151 (B and C, green) or full-length ApoE4 (D and E, green) for 24 hours. Nuclear staining with DAPI is indicated in blue with overlap images of both markers shown in either Panel C or E. Detection of 25 μg/ml nApoE41-151 was obtained using an anti-His antibody while detection of full-length ApoE4 was carried out using anti-ApoE4 N-terminal antibody. Strong nuclear localization of nApoE41-151 was evident (Panel C) following exogenous treatment, while the majority of labeling of full-length ApoE4 appeared to be cytoplasmic (Panel E). All scale bars represent 10 μm.
View Figure 1
To test whether or not FL-ApoE4 leads to the induction of inflammatory genes or other pertinent genes similar to our previously reported results for the E4-Fragment [6], we performed a transcriptome analysis of BV2 microglia cells with and without exogenous treatment with human recombinant FL-ApoE4 and identified differentially expressed genes (DEGs). We used experimental conditions identical to our previous study, including the concentration of exogenous protein. We found that treatment with FL-ApoE4 led to only 48 DEGs (fold-change > |2|, p < 0.05) (Figure 2, blue data points). This is in clear contrast with the E4-Fragment data in which 2,718 DEGs were identified as differentially expressed at the same significance level [6]. There were twenty genes with increased expression, which are listed with their functions in Table 1. Two genes stand out as obviously more up regulated (Figure 2, red circle). Gm33661 showed a 154-fold increase and Pcsk5 showed a 22.5-fold increase relative to the untreated control cells. The remaining genes were induced in the range of about 2-4-fold. There were also 28 genes with decreased gene expression. The top fifteen most down regulated are listed in Table 2. The top three down regulated genes were Il27 (-5.69-fold), Il23 (-4.00-fold), and Gdf15 (-3.69-fold). Notably, all three genes have functions involving inflammatory pathways. Moreover, of the 14 most down regulated genes of known function, 9 of these genes either play a role in the immune system or are involved in cellular growth and differentiation. This data suggests that the extracellular treatment with FL-ApoE4 actually diminishes inflammatory and growth processes in microglial cells, in contrast to previous results with the E4-fragment.
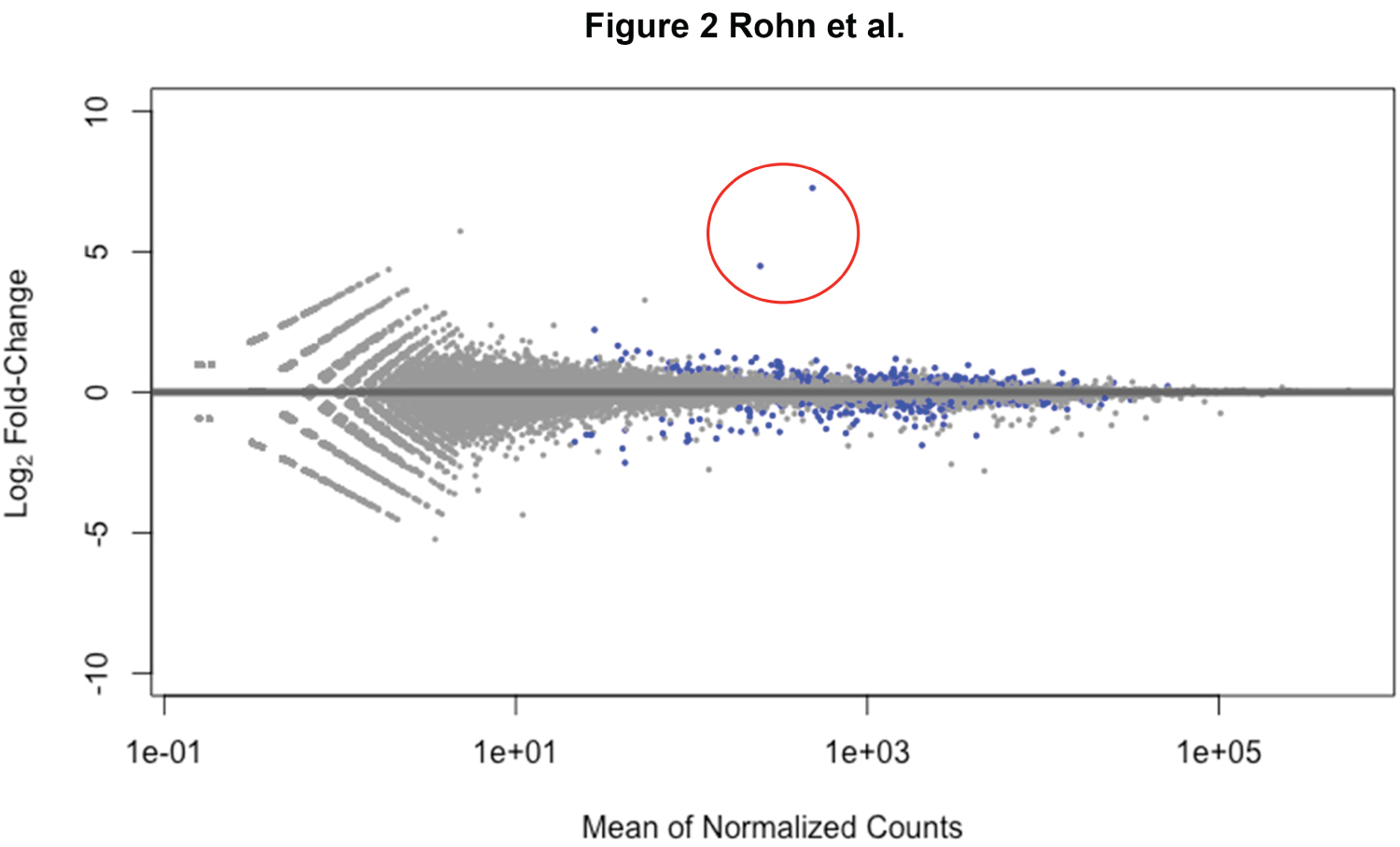 Figure 2: Few genes with large fold changes following exogenous treatment of BV2 microglial cells with full-length ApoE4. Shown is a MA-plot depicting the fold change (y-axis) over average expression for all differentially expressed genes in assessed BV2 microglia cells following FL-ApoE4 treatment at 25 μg/ml. Each log2 fold integer increment indicates a doubling of expression, with up-regulation noted as positive and down-regulation as negative. RNAseq analysis revealed that 48 genes experienced log2 fold changes > |1| when compared to the mean of normalized counts for genes from all samples. The points highlighted in blue indicate gene expression considered significant (p-values < 0.05) with two genes within the red circle exhibiting the greatest differential expression (Gm3366 and Pcsk5 at 154- and 22.5-fold increase, respectively).
View Figure 2
Figure 2: Few genes with large fold changes following exogenous treatment of BV2 microglial cells with full-length ApoE4. Shown is a MA-plot depicting the fold change (y-axis) over average expression for all differentially expressed genes in assessed BV2 microglia cells following FL-ApoE4 treatment at 25 μg/ml. Each log2 fold integer increment indicates a doubling of expression, with up-regulation noted as positive and down-regulation as negative. RNAseq analysis revealed that 48 genes experienced log2 fold changes > |1| when compared to the mean of normalized counts for genes from all samples. The points highlighted in blue indicate gene expression considered significant (p-values < 0.05) with two genes within the red circle exhibiting the greatest differential expression (Gm3366 and Pcsk5 at 154- and 22.5-fold increase, respectively).
View Figure 2
Table 1: The twenty most up-regulated genes and their functions following treatment of BV2 microglia cells with FL-ApoE4 (Greater than > 2-fold change, p < 0.05). View Table 1
Table 2: The fifteen most down-regulated genes and their functions following treatment of BV2 microglia cells with FL-ApoE4 (Greater than < 2-fold change, p < 0.05). View Table 2
In order to better understand the functional role of the differentially expressed genes, we performed a GO enrichment analysis to identify overrepresented and underrepresented biological processes in the DEGs. We used the Panther Classification System to compare our list of DEGs to the Mus musculus genome (Figure 3). We note that this analysis is difficult to interpret because there were so few differentially expressed genes. Nevertheless, the most overrepresented process was "Negative regulation of multicellular organismal processes". The underrepresented categories included "Mesoderm formation", "Endoderm formation", "Ectoderm formation" and "Negative regulation of MAP kinase activity". Notably, several processes that were enriched following treatment with the E4-fragment are not enriched in our FL-E4 data, including "inflammatory response", "activation of innate immune response", and several responses to interferons and cytokines.
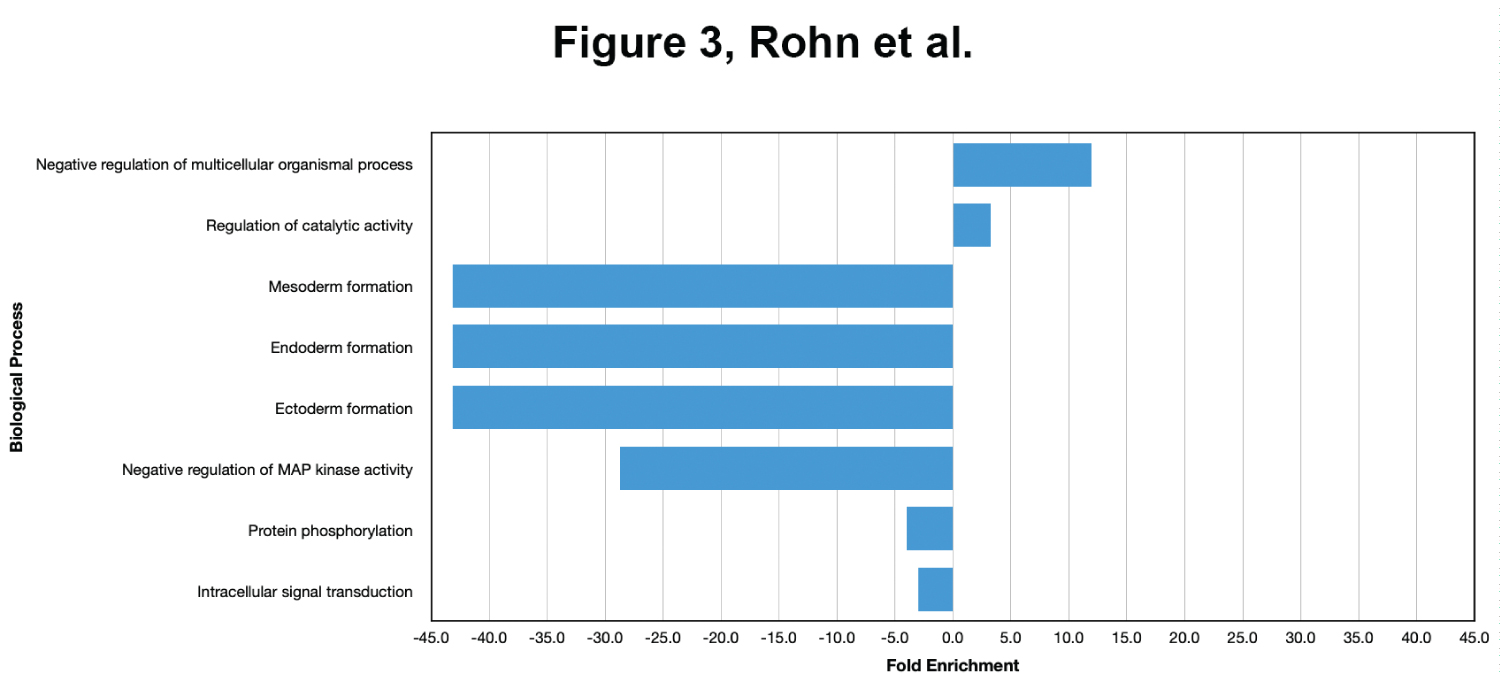 Figure 3: Enriched biological processes following treatment with full-length ApoE4. BV2 cells were plated onto 6-well plates to confluency and treated with FL-ApoE4 for 5 hours. Following treatment, total RNA was extracted and transcriptome analysis was carried out as described in the Materials and Methods. Data are expressed as fold enrichment of biological processes in BV2 microglia cells in the presence of FL-ApoE4. Up-regulated processes are involved in negative regulation of multicellular organismal processes and catalytic activity of BV2 microglia, while down-regulated processes are involved in germ layer development and cell signaling processes.
View Figure 3
Figure 3: Enriched biological processes following treatment with full-length ApoE4. BV2 cells were plated onto 6-well plates to confluency and treated with FL-ApoE4 for 5 hours. Following treatment, total RNA was extracted and transcriptome analysis was carried out as described in the Materials and Methods. Data are expressed as fold enrichment of biological processes in BV2 microglia cells in the presence of FL-ApoE4. Up-regulated processes are involved in negative regulation of multicellular organismal processes and catalytic activity of BV2 microglia, while down-regulated processes are involved in germ layer development and cell signaling processes.
View Figure 3
To further assess differences in gene expression between treatment with the FL-ApoE4 and the E4-fragment, we directly compared the DEGs from the current study with our previously published data [6]. In total, there were only 34 DEGs in the FL-E4 data that matched the DEGs in the E4-Fragment data with the same significance criteria (fold-change > |2|, p < 0.05). Table 3 lists the 10 genes with the largest increase in expression following treatment with FL-ApoE4 and their corresponding fold change following treatment with the E4-Fragment. Only four genes matched in this comparison, with one gene Slfn5 showing a much higher increase in the fragment treated cells (31.3-fold) compared to the FL-ApoE4 treated cells (2.63-fold). Table 4 shows the 10 genes most down regulated upon treatment with the FL-ApoE4 and their corresponding fold change in the E4-fragment treated cells. Surprisingly, of these 10 most down regulated genes, 9 actually showed significant up regulation in cells treated with the E4-Fragment. In addition, many of these genes showed very large increases in gene expression in the E4-Fragment treated cells. For example, Nfkbiz, Gdf15 and Il23a all showed greater than 10-fold increases, Mir155hg showed a 68.2-fold increase, and Il27 showed a 183-fold increase in the E4-fragment treated cells, but all showed ~2-5-fold decreases in the FL-E4 treated cells. Further, when analyzing all the genes down regulated by the full-length protein, we found a strong negative correlation between the fold-change in the FL-E4 data and the E4-fragment data (Pearson's r = 0.7, p < 10-16). From these reversals in the direction of gene expression, in addition to the different functional categories of DEGs, we conclude that the full-length ApoE4 protein has activity that is both diminished and opposing to the action of the E4-fragment when they are exogenously added to cultured BV2 cells.
Table 3: Fold change of the Top 10 most up-regulated genes following treatment of BV2 microglia cells with FL-ApoE4 and their fold change following treatment with nApoE41-151 (p < 0.05). View Table 3
Table 4: Fold change of the Top 10 most down-regulated genes following treatment of BV2 microglia cells with FL-ApoE4 and their fold change following treatment with nApoE41-151 (p < 0.05). View Table 4
The exact mechanism by which the ApoE4 protein enhances dementia risk has been extensively investigated and the general consensus to date is ApoE4 is less effective in promoting the clearance of beta-amyloid deposits, one of the hallmark molecular signatures of AD [12]. In this regard, both pathological and neuroimaging studies have indicated that beta-amyloid deposition occurs to a greater extent and earlier in the disease process in APOE4 carriers [13-15]. However, alternative mechanisms have recently come to light including accumulating evidence that ApoE4 is more susceptible to proteolytic cleavage and amino-terminal fragments obtain a toxic-gain of function [16]. In support of this hypothesis is the presence of major ApoE4 fragments in ApoE4 transgenic mice [17] as well as in the human AD brain [5,18,19].
In a previous study, we reported that an amino-terminal fragment of ApoE41-151 (E4-Fragment) is taken up by microglia cells, traffics to the nucleus and changes gene expression culminating in cellular toxicity and an M1 activated phenotype [5,6,20]. In addition, it was shown that while the E4-Fragment can specifically bind to the TNF-α promoter region and causes significant toxicity, FL-ApoE4 was without effect in either regard under identical experimental conditions [5,20]. These data suggest a novel role of fragmented ApoE4 beyond lipid metabolism. In the present study, we examined whether this function extends to FL-ApoE4 or is specific to the E4-Fragment.
To assess this possibility, we performed a transcriptome analysis in BV2 microglia cells following exogenous treatment with FL-ApoE4 and compared the results to E4-Fragment previously reported [6]. Treatment with FL-ApoE4 led to a much lower impact on gene expression. Indeed, a total of only 48 DEGs (fold > 2, p < 0.05) were identified for FL-ApoE4 as compared to 2,718 DEGs for the E4-Fragment. Specifically, the top 3 genes induced by FL-ApoE4 were Gm33661 (154-fold), Pcsk5 (22.5-fold), and Serpina3f (4.66-fold). Gm33661 was by far the most significantly up regulated gene but unfortunately, very little is known about this predicted gene. Although no known function has been assigned to Gm33661, bioinformatics analysis indicates that this putative gene most likely represents a long non-coding RNA (lncRNA). In an effort to obtain additional information about this gene, we employed the use of the SEEKR algorithm that used relative frequencies of short motifs called k-mers to infer function based on similarity to other lncRNAs [21,22]. K-mer profiles were tested for similarity using Pearson correlations. The output of such an analysis indicated that the majority of putative functions that align with Gm33661 fall under the categories of maintenance of pluripotency and regulation of nervous system development through methylation. In this regard, Gm33661 likely is down regulated during differentiation (Pearson correlation > 0.9).
Pcsk5, on the other hand is a well characterized gene having major roles in lipoprotein metabolism [23]. Interestingly, a recent genome-wide association study (GWAS) identified single nucleotide polymorphisms (SNPs) linked to cognitive impairment and hypotension [24].
The induction of Serpina3f by FL-ApoE4 is of significance as this gene is a member of the serpin family of proteins, a group of proteins that inhibit serine proteases. This gene is one in a cluster of Serpin genes located on the q arm of chromosome 14. Another member of this class, Serpina3g was also significantly up regulated by FL-ApoE4 (2.71-fold) (Table 1). Variations in the Serpina3 gene have been implicated in AD affecting both age-at-onset and disease duration [25]. Molecularly, it has recently been shown that Serpina3 SNPs can prolong toxic oligomeric forms of beta-amyloid leading to neuronal cell death, in vitro [26].
Overall, it appeared that FL-ApoE4 was a stronger suppressor of gene expression. This seemed to be the case for enrichment pathways. In terms of enriched pathways, strong negative-fold enrichment was observed for tissues involved in germ layer development including mesoderm, endoderm, and ectoderm. This may be linked to the down regulation of several members of the Dusp gene family including Dusp1, 2, 4, 5, 8, 14, and 18 (the raw data file can be accessed through supplemental data file, S1). The Dusp proteins belong to the extended family of tyrosine phosphatases, whose major targets are the map kinases [27]. It has been postulated that Dusp's regulation of cytokines may serve as a mechanism to dampen down immune responses [28]. These results are consistent with previous findings using isolated astrocytes from human AD post-mortem samples [29]. A transcriptome analyses of these astrocytes in which subjects were ApoE4+ revealed a down regulation of a number of genes associated with map kinase signaling, including Dusp19 [29].
In a final analysis, we compared FL-ApoE4 RNAseq data with those obtained under identical experimental conditions following treatment with the E4-Fragment [6]. Of the top 10 genes upregulated by FL-ApoE4, only 4 genes matched with the E4-Fragment: Serpina3f and 3g, Slfn5, and Siglece. Interestingly, both FL-E4 (4.66-fold) and the E4-Fragment (4.85-fold) led to a similar up regulation of Serpina3f. As previously stated, the family of Serpina3 genes has been linked to AD [25]. Our results support those of a recent study that demonstrated the APOE4 gene was associated with up regulation of multiple Serpina3 genes including Serpina3f [30].
Perhaps the strongest contrast between FL-ApoE4 and the E4-Fragment were those top genes down regulated by FL-ApoE4. The significant negative correlation indicates that in general, genes down regulated by FL-ApoE4 tended to be up regulated by the E4 fragment. Of the 10 top genes down regulated by FL-ApoE4, 9 were conversely significantly up regulated by E4-Fragment (Table 4). The top three genes, Il27, Il23a, and Gdf15 all have important functions in inflammatory responses as either cytokines or a ligand that regulates the TGF-α. In addition, growth differentiation factor 15 (GDF15) has recently been shown to be a risk factor for AD and cognitive decline [31]. These data suggest opposing actions of these two proteins with FL-ApoE4 appearing to dampen inflammatory pathways in contrast to the E4-Fragment which is a strong activator.
Herein we report new findings examining the effects of the ApoE4 protein on gene expression. Our results support the notion that the linchpin linking the APOE4 gene to dementia risk may involve not the full-length form of the protein itself, but an amino-terminal fragment that is a potent activator of inflammatory pathways following exposure to microglia cells. Collectively, these results now provide additional support at the gene level that amino-terminal fragments of ApoE4 result in a toxic-gain of function. These data have important implications in that the neutralization of this amino-terminal fragment of ApoE4, specifically, may serve as an important therapeutic strategy in the treatment of AD.
This work was funded by National Institutes of Health Grant 2R15AG042781-02A1. The project described was also supported by Institutional Development Awards (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under Grants #P20GM103408 and P20GM109095. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We also acknowledge support from the Biomolecular Research Center at Boise State with funding from the National Science Foundation, Grants #0619793 and #0923535; the MJ Murdock Charitable Trust; and the Idaho State Board of Education. Analyses in this publication were supported by NSF Track 2 EPSCoR Program under award number OIA-1826801.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
TR designed experiments, helped construct tables and figures, and wrote the manuscript. JB, TP, and SG analyzed data, constructed figures, and contributed to writing of the manuscript. NI helped carry out experiments. TarunS, TonyaS, and AK helped analyze data. EH helped analyze data, provided input and resources for experiments, and contributed to writing of the manuscript.