The attempted healing of tendon after acute injury (overloading, partial tear or complete rupture) proceeds via the normal wound healing cascade involving hemostasis, inflammation, matrix synthesis and matrix remodeling. Depending on the degree of trauma and the nature of the post-injury milieu, a variable degree of healing and recovery of function occurs. Post-injury analgesia is often achieved with NSAIDs such as Ibuprofen, however there is increasing evidence that NSAID usage may interfere with the healing process. This study aimed to investigate the cellular mechanism by which IBU therapy might lead to a worsening of tendon pathology.
We have examined the effect of oral Ibuprofen, on Achilles tendon healing in a TGFb1-induced murine tendinopathy model. Dosing was started 3 days after initial injury (acute cellular response phase) and continued for 22 days or started at 9 days after injury (transition to matrix regeneration phase) and given for 16 days. Cellular changes in tendon and surrounding peritenon were assessed using Hematoxylin/Eosin, chondroid accumulation with Safranin O and anti-aggrecan immunohistochemistry, and neo-vessel formation with GSI Lectin histochemistry. Markers of inflammation included histochemical localization of hyaluronan, immunohistochemistry of heavy chain 1 and TNFα-stimulated glycoprotein-6 (TSG6). Cell responses were further examined by RT-qPCR of 84 NFκB target genes and 84 wound healing genes. Biomechanical properties of tendons were evaluated by tensile testing.
At a clinically-relevant dosage, Ibuprofen prevented the process of remodeling/removal of the inflammatory matrix components, hyaluronan, HC1 and TSG6. Furthermore, the aberrant matrix remodeling was accompanied by activation at day 28 of genes (Col1a2, Col5a3, Plat, Ccl12, Itga4, Stat3, Vegfa, Mif, Col4a1, Rhoa, Relb, F8, Cxcl9, Lta, Ltb, Ccl12, Cdkn1a, Ccl22, Sele, Cd80), which were not activated at any time without the drug, and so appear most likely to be involved in the pathology. Of these, Vegfa, Col4a1, F8, Cxcl9 and Sele, have been shown to play a role in vascular remodeling, consistent with the appearance at 25 days of vasculogenic cell groups in the peritenon and fat pad stroma surrounding the Achilles of the drug-dosed mice. Tensile stiffness (p = 0.004) and elastic modulus (p = 0.012) were both decreased (relative to age-matched uninjured and non-dosed mice) in mice dosed with Ibuprofen from day 3 to day 25, whether injured or not.
We conclude that the use of Ibuprofen for pain relief during inflammatory phases of tendinopathy, might interfere with the normal processes of extracellular matrix remodeling and cellular control of expression of inflammatory and wound healing genes. It is proposed that the known COX2-mediated anti-inflammatory effect of ibuprofen has detrimental effects on the turnover of a pro-inflammatory HA matrix produced in response to soft-tissue injury, thus preventing the switch to cellular responses associated with functional matrix remodeling and eventual healing.
TGF-β1: Transforming Growth Factor beta 1; IBU: Ibuprofen; ECM: Extracellular Matrix; UI: Uninjured; COV: Coefficient of Variation; NSAIDs: Non-Steroidal Anti-inflammatory Drugs; HA: Hyaluronan; HC1: Inter-alpha-trypsin Heavy Chain 1; EMT: Epithelial Mesenchymal Transition; IaI: Inter-alpha-trypsin Inhibitor
Tendinopathy, NSAID, Inflammation, Wound healing, Hyaluronan, Angiogenesis, Biomechanical properties
The Achilles tendon is the largest tendon in the body. Its primary function involves push-off and maintaining the overall position of the tibia during gait. Tendinopathy of the Achilles is a major health care concern with specific focus to orthopaedic foot and ankle surgeons. It often presents as pain and swelling within the tendon proper, and has been found to be associated with obesity, hypertension, and steroid use [1], each of which have shown an end-organ effect of a decrease in local microvascularity. While this correlation is consistent with a role for hypoxia in the development of Achilles tendinopathy [2], effective therapies to alleviate symptoms remain limited. Current treatment protocols frequently include non-steroidal anti-inflammatory drugs (NSAIDs such as Ibuprofen) in combination with activity modification, rest, physical therapy and various bracing modalities. Surgical options do exist however they involve significant compliance during recovery and may lead to worsening morbidity related to wound complications or improper healing (reviewed in [3]). While NSAIDs provide short-term relief of pain through inhibition of cyclooxygenases-1 and -2, (and thereby inhibition of prostaglandin production), it is also well established that NSAIDs modulate multiple targets and can have multiple molecular effects apparently unrelated to cyclooxygenase inhibition [4]. These effects include binding to membrane transporters, such as hPEPT1 [5], enhancing microtubule formation in epithelial cells [6], modulation of cell membrane topography [7], inhibition of caspases [8] and induction of apoptosis [9]. NSAIDs have also been implicated in cardiac pathology [10] and there is mounting evidence that ibuprofen may also interfere with tendon repair [11-13].
We aimed to describe the effect of IBU therapy on tendon pathological features in a murine model of tendinopathy, and to provide mechanistic details of the cellular responses involved.
Unilateral tendinopathy was induced in the Achilles tendon of skeletally mature male C57Bl/6 mice by injections, on days 0 and 2, of 100 ng of active rhTGF-β1 (human recombinant, Peprotech, Inc., Rocky Hill, NJ) into the mid-portion of the tendon [2,14-16]. The injections were given between 2 pm - 4 pm and all euthanasia and tissue collections were between 9 am - 11 am. Mice had free access to food and drinking water and were allowed normal cage activity.
Ibuprofen (IBU) was administered orally as described by Ezell, et al. [17]. Briefly, a pediatric IBU oral suspension (20 mg/mL, CVS Pharmacy) was diluted with purified water to a final concentration of 1 mg/mL. Drinking bottles were wrapped in silver foil and the drug was renewed every 5 days. Drug was either initiated at 3 days after initial injury (acute cellular response phase) and administered for 22 days or initiated at 9 days after injury (matrix regeneration phase) and administered for 16 days. Average water consumption was ~3.8 mL (~3.8 mg IBU) per mouse per day. Experimental groups, outcome measures, and mouse numbers for each, are summarized in Supplementary Table 1. Mice were weighed before induction of tendinopathy and again before euthanasia, and no marked difference in average body weight was observed between experimental groups. All animal use was approved by the IACUC of Rush University.
Following euthanasia, Achilles tendons (including peritenon) were immediately isolated from all surrounding tissue, placed in RNA. Later and stored at -20 ℃. RNA was isolated from each pool of tissue generated for each experimental group (Supplementary Table 1), as previously described [14]. Briefly, pooled tissue was fragmented under liquid nitrogen in a Bessman Tissue Pulverizer and extracted in 1 mL of Trizol by vortexing for 60 seconds. RNA purification was done with the RNeasy MiniKit (Qiagen, Cat #: 74104, Valencia, CA), and yields were between 300-500 ng RNA per tendon. RNA quality (A260:A280) was > 1.90 for all preparations, and 2 ug of each preparation was used for cDNA synthesis with the RT2 First Strand Kit (Qiagen). mRNA abundance for 84 NFκb target genes and 84 wound healing genes were determined using SYBR qt-PCR array plates (PAMM-121A, PAMM-225ZA Qiagen, Valencia, CA). Alphabetic gene listings are shown in Supplementary Table 2 and Supplementary Table 3. The reproducibility of the QPCR array assay was confirmed as described previously [3,18]. It should be noted that data for individual pools represents the average expression in 14-20 individual tendons (Supplementary Table 1). Changes in transcript abundance (ΔCt = Ct for transcript of interest minus Ct for the housekeeping gene, B2m) were used to calculate the fold change (2^-ΔΔCt) relative to un-injured levels for each experimental and control group. A 1-way ANOVA with Tukey's post-hoc tests was conducted using GraphPad Prism 7 (La Jolla, CA) on the ΔCt values to determine the significance (p < 0.05) of differences in the expression of genes. Specifically, 25 d post-injury data was compared to UI data and either E-IBU or L-IBU data.
Supplementary Table 4: Mean mRNA Abundance of individual genes in UI pools (ND* = Not detectable, ΔCt > 14.0). View Supplementary Table 4
Ankle joints (n = 3 per experimental group) were processed as previously described [14,15]. Briefly, joints were fixed in formaldehyde, decalcified in EDTA, processed, embedded in paraffin, and 5 µm thin sagittal sections cut through the entire joint. Sections (n = 36) from the mid-portion of each specimen, which included the Achilles-calcaneus insertion and the bursa were stained as follows: For histopathological evaluation of tissue responses to injury and drug treatment, section numbers 1,7,13,20,25,31 were stained with Hematoxylin/Eosin [14] and section numbers 2,14,21,26,32 were stained with Safranin-O (SafO) [15]. The remaining sections were used for immunohistochemistry: Briefly, sections (n = 6 per antibody) were deparaffinized and incubated overnight at 4 ℃ with the following probes: anti-TSG6 (5 ug/mL [19]), anti- Inter-alpha-trypsin Heavy Chain 1 (HC1) (1 ug/mL [20]) or aggrecan-DLS (1 ug/mL [15]), followed by biotinylated anti-rabbit IgG as secondary antibody. HA (hyaluronan) was localized with biotinylated HABP [18]. Neovascularization in the peritenon and the fat pad was assessed by immunochemistry with GSI Lectin (10 ug/mL; Vector Laboratories, Inc. Burlingame, CA 94010) [21]. All sections were counterstained with methyl green. Negative control staining is shown in Supplementary Figure 1.
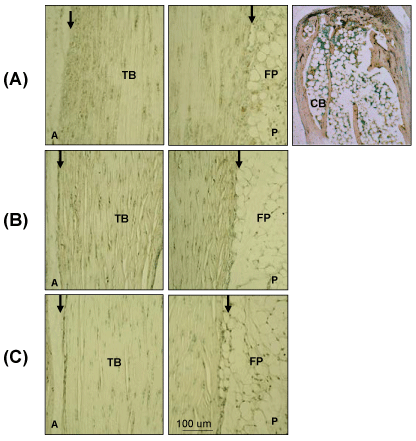 Supplementary Figure 1: Negative control Images for IHC and Immunochemistry.
Supplementary Figure 1: Negative control Images for IHC and Immunochemistry.
Sections were treated (A) with non-immune rabbit serum, following Proteinase K treatment, (B) without GSI Lectin or (C) biotinylated HABP following Streptomyces Hyaluronidase digestion of HA. TB: Tendon Body; FP: Fat Pad; P: Posterior; CB: Calcaneus Bone; A: Anterior; Peritenon regions are indicated by black arrows. The staining of the cancellous bone represents non-specific staining due to binding of the secondary antibody in proteinase K pretreated samples.
View Supplementary Figure 1
Tensile testing and analysis of mechanical properties were carried out as described by Wang, et al. [22]. Briefly, the Achilles/calcaneus complex was dissected from the surrounding tissues, followed by measurements of tendon width and thickness, from which cross-sectional was computed. The foot was potted in dental cement and the tendon-bone construct was mounted onto a materials testing system (MTS Insight 10, Eden Prairie, MN). Tensile testing (in a saline-filled chamber) consisted of preconditioning followed by a load to failure test at 0.05 mm/sec. 1-way ANOVA and Tukey's Multiple Comparison Tests were performed using JMP (SAS, Cary, NC) for statistical analysis of the data obtained from the 5 experimental groups listed in Supplementary Table 1.
Table 1: Fold Change expression (relative to UI) of Wound Healing genes after injection of TGF-β1. View Table 1
Low magnification SafO of the ankle joint (Supplementary Figure 2A) showed that by 3 d there was hyperplasia of both the anterior peritenon and fat pad and the posterior peritenon along with swelling of the tendon body. By 9 d, remodeling of the tendon ECM was evident as shown by chondroid deposition. At 25 d, there was evidence for reversal and repair as suggested by chondroid removal and the essentially normal appearance of both the peritenon and fat pad. The pattern of chondroid deposition in the model was confirmed by IHC localization of aggrecan-positive cells (Supplementary Figure 2B). Transient accumulation of neutrophils in vascularized regions of the fat pad were also observed in this acute phase response to the TGFb1 injury (Supplementary Figure 3).
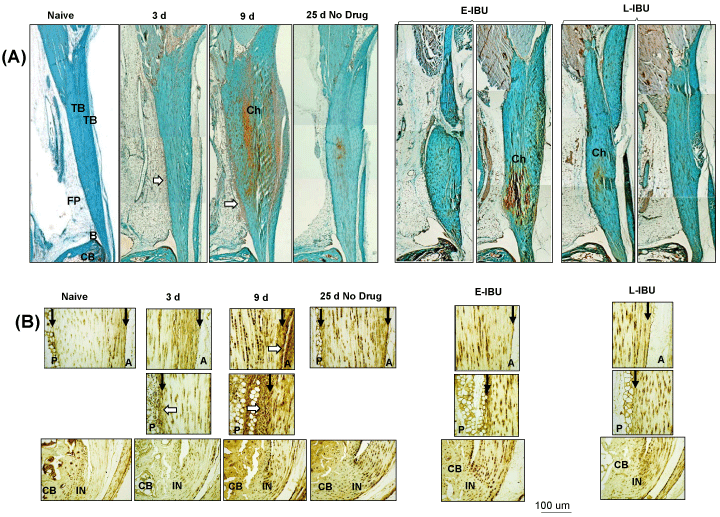 Supplementary Figure 2: SafO staining (A) and Aggrecan IHC (B) of the Achilles tendon and surrounding tissues, pre- and post-injury and after IBU dosing.
Supplementary Figure 2: SafO staining (A) and Aggrecan IHC (B) of the Achilles tendon and surrounding tissues, pre- and post-injury and after IBU dosing.
Panel A: Sagittal sections were stained as described in the Methods. TB: Tendon Body; FP: Fat Pad; B: Bursa; CB: Cancellous Bone; Ch: Chondroid; P: Posterior; A: Anterior; IN: Insertion site. Areas of increased cellularity of the periosteum and adipose stroma are indicated by white arrows. Anterior and Posterior peritenon regions are indicated by black arrows.
View Supplementary Figure 2
 Supplementary Figure 3: Altered cellularity in posterior Peritenon and fat pad following injury and after IBU treatment.
Supplementary Figure 3: Altered cellularity in posterior Peritenon and fat pad following injury and after IBU treatment.
Sagittal sections were stained as described in the Methods. TB: Tendon Body; FP: Fat Pad; N: Neutrophils; BV: Blood vessel; black arrows show the peritenon regions. Accumulation of neutrophils in the fat pad stroma in 3 d samples, hyperproliferated stromal cells at 9 d and persistent accumulation of hyper-cellularity in the adipose stroma after IBU treatment are marked by black arrow heads. A high magnification of these cells is shown in panel B.
View Supplementary Figure 3
To more closely define ECM changes in the inflammatory response to the TGFb1 injury, we performed IHC localization of components of the well-characterized inflammatory HA matrix [22-25], typically containing HA (Figure 1), HC1 (Figure 2) and TSG6, which transfers HC1 from bikunin onto HA [25] (Figure 3). HA staining increased between 3 d and 9 d, together with HC1 and TSG6, and this was evident in the tendon body, insertion sites, the peritenon and adipose stroma. By d 25 however, HA and HC1 reactivity was greatly diminished, indicating a clearance of this pro-inflammatory matrix most likely by resident phagocytic cells, including macrophages [27,28]. In contrast, TSG6 protein remained elevated in tendon cells, even at d 25 post-injury, however this did not appear to maintain a high level of HC1-HA matrix, presumably due to a lack of substrate HA or Inter-alpha-trypsin inhibitor (IaI), or inactivation of the transferase activity of the TSG6.
 Figure 1: Histochemical localization of HA in Achilles tendon and surrounding tissues following TGFb1 induced injury.
Figure 1: Histochemical localization of HA in Achilles tendon and surrounding tissues following TGFb1 induced injury.
Sections were treated and stained with HABP as described in the Methods. TB: Tendon Body; FP: Fat Pad; CB: Cancellous Bone; P: Posterior; A: Anterior; IN: Insertion site. Peritenon regions are indicated by black arrows and the space bar in the bottom panel = 100 µm.
View Figure 1
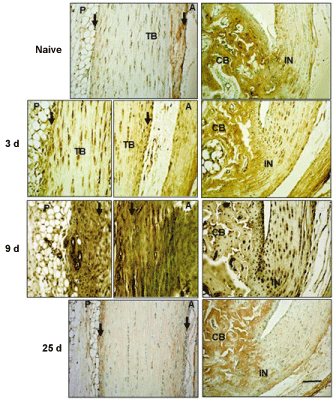 Figure 2: Immunohistochemical localization of HC1 in Achilles tendon and surrounding tissues.
Figure 2: Immunohistochemical localization of HC1 in Achilles tendon and surrounding tissues.
Sections were stained with anti-HC1 antibodies following Proteinase K treatment for antigen retrieval, as described in the Methods. TB: Tendon Body; FP: Fat Pad; CB: Cancellous Bone; P: Posterior; A: Anterior; IN: Insertion site. Peritenon regions are indicated by black arrows, and the space bar in the bottom panel = 100 µm. The staining of the cancellous bone represents non-specific staining due to binding of the secondary antibody in proteinase K pretreated samples.
View Figure 2
 Figure 3: Immunohistochemical localization of TSG6 in Achilles tendon and surrounding tissues.
Figure 3: Immunohistochemical localization of TSG6 in Achilles tendon and surrounding tissues.
Sections were stained with anti-TSG6 antibody, as described in the Methods. TB: Tendon Body; FP: Fat Pad; CB: Cancellous Bone; P: Posterior; A: Anterior; IN: Insertion site. Peritenon regions are indicated by black arrows, and the space bar in the bottom panel = 100 µm.
View Figure 3
The expression of genes in Achilles tendons (including peritenon) from naïve mice and at 3 d, 9 d and 25 d after injection of TGFb1 were determined. For each array (Table 1 and Table 2), the genes are arranged alphabetically under each pathway group and the values show the fold-change in expression at each time point, relative to UI levels.
Table 2: Fold Change expression (relative to UI) of Nfkb Target genes after injection of TGF-β1. View Table 2
Wound healing pathway genes (Table 1) which were > 5-fold activated at 3 d included ECM structure genes (Col5a1), ECM remodeling factors (Timp1, Mmp9, Serpine1), cell adhesion proteins (Itga5, Cdh1), growth factors (Hbegf, Hgf, Wisp1) and chemokines (Ccl7, Cxcl5). NFκb target genes > 5-fold activated at 3 d (Table 2) included chemokines and cytokines (Tnf, Ilr2, Cxcl1, Cxcl3, Il1b, Il6), acute inflammatory response proteins (Ptgs2, Tnfrsf1b, Myd88), a stress factor (Plau), an NFκb signaling factor (Rel) and an apoptosis factor (Nr4a2). Of the 23 genes activated > 5-fold at 3 d, all but three (Col5a1, Mmp9 and Tnf) were still activated at 9 d (Table 1 and Table 2) but returned close to naïve expression levels by 25 d, consistent with the histological changes indicating a resolution of the inflammatory wound healing response at that time (Figure 1, Figure 2 and Supplementary Figure 2). Notably, a different group of 21 genes (Col14a1, Col1a1, Col3a1, Col5a2, Ctsk, Mmp2, Mmp9, Plaur, Igf1, Il10, Tnf, Ccl5, Cxcl10, Il12b, Il1rn, Ccr5, C4a, Bcl2a1a, Vcam1, Cd83) were more highly expressed at 9 d than at 3 d and for 10 of these (Col14a1, Col1a1, Mmp2, Igf1, Il10, Ccl5, Il12b, C4a, Bcl2a1a, Vcam1, Cd83) the initial activation was even delayed until 9 d, consistent with a primary role for these genes in the transition from acute inflammation (3 d) to tissue recovery (25 d). Thus, the initial increase in expression of Col14a1, Col1a1, Col3a1, Col5a1, Col5a2 and Igf1 at 9 d would be expected to promote repair of the collagenous and proteoglycan matrix respectively, and normalization of expression of these genes at 25 d indicates that the repair was largely complete, much as observed histologically (Figure 1, Figure 2 and Supplementary Figure 2). In contrast, seven genes (Il1b, C4a, Ptgs2, Nr4a2, Vcam1, Mmp2 and Cdh1) remained greater than 5-fold activated at 25 d, suggesting that the initial inflammatory response, indicated by high expression of Il1b and Ptgs2, was not entirely resolved even at 25 d, also consistent with the high TSG6 protein at this time (Figure 3).
A consideration of established functions, suggests that at least some of the 23 genes activated > 5-fold on 3 d are apparently involved in development of the 9 d pathology (tissue swelling, chondroid deposition, inflammatory HA matrix deposition). For example, Serpine1 and Timp1 can be broadly considered as markers of fibrosis [28,29] whereas activation of Cdh1 (E-cadherin) suggests epithelial-mesenchymal transitional (EMT) activity [31] in the peritenon. Furthermore, activated expression of Il1b and Il1r2 indicates both a pro-inflammatory response to the injury and its local control by epithelial cells of the peritenon [32-33]. Activation of Il6 (~142-fold) at 3 d is consistent with a broad effect of the injury on immune and non-immune cells with both, pro- and anti-inflammatory properties. In addition, the highly activated and coordinated expression of Cxcl1 (~28-fold), Cxcl3 (~137-fold) and Cxcl5 (~510-fold) supports the occurrence of the pro-inflammatory response that has been reported after IL1β treatment of MSCs [34].
The expression of other genes (Il15, Il4, Tnfsf10, Il2ra, F3, Agt, Ncoa3, Fas, Sod2, Map2k6, Mitf, Nqo1, Itgb6, Actc1, Cxcl11, and Fgf10) was inhibited greater than 5-fold at 3 d and all were essentially normalized by 25 d. A putative function for these genes in the injured tendon is not obvious, however in two cases where the genes have been shown to be protective (Il4 [35] and Sod2 [36]) the inhibition of expression would be expected to enhance the general inflammatory response.
When compared to no drug, E-IBU had marked effects on the histological appearance of tendon and peritenon at 25 d. Tendons in dosed mice retained elevated levels of chondroid in the tendon body (Supplementary Figure 2A and Supplementary Figure 2B), indicating that E-IBU interfered with reparative removal of the accumulated post-injury ECM. Most notable however was the increased abundance of blood vessels in the adjacent fat pad (Supplementary Figure 3A) in both E-IBU and L-IBU groups at 25 d. The increased angiogenic response of fat pad stromal cells was confirmed by GSI lectin histochemistry (Figure 4), which has been used to assess formation of new blood vessel at sites of injury [3]. Without IBU dosing (Figure 4A) a minor and transient increase in small vessels was seen in the fat pad (white arrows) at 9 d, but no such structures were detectable at 25 d. By comparison, multiple groups of strongly GSI-positive cells were seen in the fat pads of both E-IBU and L-IBU groups (Figure 4B, white arrows). However, since the GSI-positive cells had not formed tubular structures, as expected of neo-vessels, they may represent either precursor neo-vessels, or partially differentiated multi-potent adipose-derived stromal cells, which can exhibit the 'vascular mimicry' which often accompanies epithelial-mesenchymal transition [37].
 Figure 4: Histochemical Staining with GSI Lectin of the Achilles tendon and surrounding tissues following TGFb1 induced injury without (A) and with (B) IBU treatment.
Figure 4: Histochemical Staining with GSI Lectin of the Achilles tendon and surrounding tissues following TGFb1 induced injury without (A) and with (B) IBU treatment.
Sections were treated and stained with GSI Lectin, as described in the Methods. TB: Tendon Body; FP: Fat Pad; P: Posterior; A: Anterior; Peritenon regions are indicated by black arrows and the space bar in the bottom panel = 100 µm. White arrows indicate the stained cell groups (indicative of vasculogenic mimicry) that accumulated in the fat pad stroma at 9 d post-injury and at 25 d in E IBU and L IBU groups.
View Figure 4
Additional histological differences between IBU-dosed and non-dosed specimens were seen after staining for HA and HC1 (Figure 5A and Figure 5B, respectively). In the non-dosed samples, HA and HC1 reactivity had been largely restored to low pre-injury levels by 25 d. This is likely through clearance of the pro-inflammatory matrix by resident phagocytic cells, including tissue macrophages [27], However, in both E-IBU and L-IBU groups, HA (Figure 5A) and HC1 (Figure 5B) staining remained elevated in the tendon body and throughout the peritenon, indicating that the drug dosing impaired or delayed the clearance of this pro-inflammatory matrix, and therefore prolonged inflammation and ECM remodeling by resident cells.
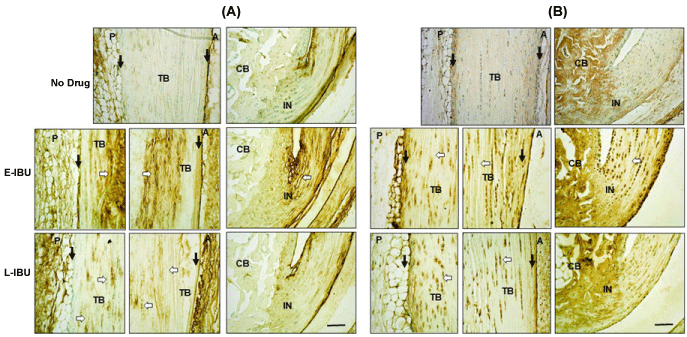 Figure 5: Accumulation of HA (A) and HC1 (B) in the tendon body and the peritenon at 25 d with IBU treatment.
Figure 5: Accumulation of HA (A) and HC1 (B) in the tendon body and the peritenon at 25 d with IBU treatment.
Sections were stained as described in the Methods. TB: Tendon Body; CB: Calcaneus; IN: Insertion; P: Posterior; A: Anterior; Peritenon regions are indicated by black arrows and the space bar in the bottom panel = 100 µm. White arrows highlight ECM regions of the tendon body with persistent HA accumulation after IBU treatment (A), and persistence of HC1 positive tendon and at 25 d in E-IBU and L-IBU groups (B).
View Figure 5
At the histological level, IBU dosing resulted in increased levels of chondroid and HA/HC1 in the tendon as well as an apparent attempt at neovascularization of the fat pad stroma. Taken together, this suggested that the drug interfered with the capacity of cells to modulate inflammation associated pathways and to achieve clearance via remodeling of the post-injury generated ECM. To provide mechanistic insight we compared expression levels of Wound healing and NFκb target genes in tendons from IBU-dosed and non-dosed mice (Table 3). This analysis revealed that E-IBU resulted in a higher (p < 0.05) expression (relative to 25 d no drug), for 26 NFκb target genes and 24 wound healing genes, whereas L-IBU enhanced expression of only 11 and 6 genes respectively. This suggested that for most genes the drug-enhanced expression seen at 25 d was dependent on IBU administration starting in the acute inflammatory post-injury period (at 3 d). Since the relatively low expression at 25 d without IBU (Table 1 and Table 2) followed normalization from stimulated levels at 3 d and 9 d, the elevated levels at 25 d (relative to no dosing) would be consistent with a drug-mediated inhibition in the normalization process. If the apparent loss of clearing capacity with IBU dosing (i.e. accumulated chondroid, vascular adipose, GSI-positive cells and HA/HC1 matrix) results from the enhanced expression of genes (Table 3), this implicates up to 50 genes in the IBU-induced pathology. Of the 50 genes, 25 were activated by injury (relative to naïve) at 3 d and/or 9 d without drug (Table 1 and Table 2) and were also enhanced (relative to no drug) at 25 d by IBU dosing.
Table 3: Fold Change expression (relative to UI) of Nfkb Target genes after injection of TGF-β1. View Table 3
Genes that were not activated at any time in the no drug group, but enhanced by E-IBU appear most likely to have been responsible for the IBU-induced changes in cell responses and downstream pathology. Notably all of those have been reported to play a role in vascular remodeling (Vegfa [38], Col4a1 [39], F8 [40], Cxcl9 [41], Sele [42]) consistent with the appearance at 25 d of vasculogenic cell groups in the peritenon and fat pad of the IBU-dosed mice.
It is worth noting that those genes where expression was inhibited (> 5-fold), rather than activated, at 3 d were essentially normalized by 25 d with no drug, and were not markedly affected by IBU dosing.
Notably, it was shown that IBU dosing of naïve mice for 22 days caused a 25-30% loss in stiffness and elastic modulus, suggesting that a low-level of PGE2-mediated inflammation is essential for normal homeostatic maintenance of tendon mechanics. However, IBU dosing also activated genes such as u-PAR (Plaur) which has been linked to tendon healing [43], Itga5 and RhoA which have been linked to repair [44,45] and Cd40lg associated with inflammation control [46]. It is unknown how activation of these genes might lead to the observed loss of stiffness or elastic modulus in such tendons (Figure 6 and Supplementary Figure 4).
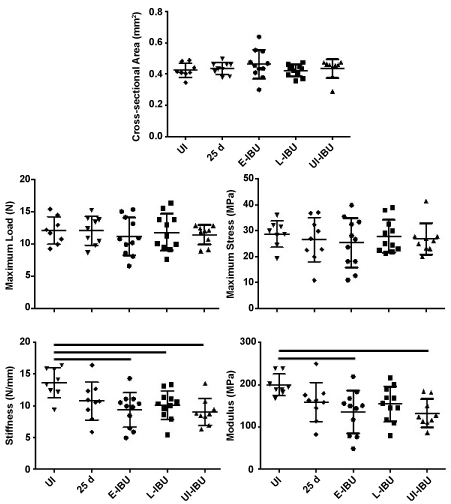 Figure 6: Effect of IBU administration on Achilles tendon mechanical properties.
Figure 6: Effect of IBU administration on Achilles tendon mechanical properties.
The scatter plots show data for individual tendons in each group. For each group, horizontal lines denote mean ± one standard deviation. Horizontal lines above the plots denote statistically significant differences between groups.
View Figure 6
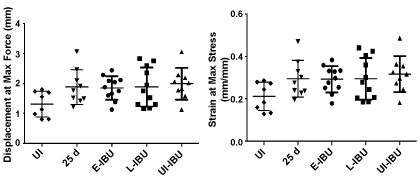 Supplementary Figure 4: Effect of IBU administration on Achilles tendon mechanical properties.
Supplementary Figure 4: Effect of IBU administration on Achilles tendon mechanical properties.
The scatter plots show data for individual tendons in each group. For each group, horizontal lines denote mean ± one standard deviation.
View Supplementary Figure 4
Tensile stiffness (p = 0.004) and elastic modulus (p = 0.012) were both decreased (relative to age-matched uninjured and non-dosed mice) in mice dosed with IBU from 3 d to 25 d, whether injured (E-IBU) or not (UI-IBU) (Figure 6). This effect of IBU appears to be independent of the changes in wound healing and NFκb gene expression in injured samples (Table 3) since those changes required TGFb1 injection. No statistically significant differences were noted among the experimental groups with regard to cross-sectional area (p = 0.56), maximum load (p = 0.90), or maximum stress (p = 0.89). Additionally, no statistically significant differences were found for displacement at maximum force (p = 0.09) and strain at maximum stress (p = 0.10) (Supplementary Figure 4), although a statistical trend towards increases in these parameters with injury (with and without IBU) may contribute to the significant changes in stiffness and elastic modulus, respectively. Notably, the coefficient of variation (COV) for both geometric and mechanical properties was generally lowest for the UI-IBU group, while the COV of the E-IBU group was consistently higher than that of the L-IBU group, particularly for material (in comparison to structural) properties.
This study was motivated by the question of how dosing with NSAIDs such as IBU for pain relief, might influence the pathogenesis of tendinopathies [11-13]. For this purpose we studied the histopathological, biochemical responses and biomechanics of the Achilles tendon in a non-surgical model of tendinopathy in mice. Using such outcome measures, a detailed characterizing of phases of acute post-injury responses (1-2 weeks) followed by a recovery and repair period (2-4 weeks) has been reported [3,14,15].
The data obtained in this study reveals that IBU dosing, especially when started during the acute injury response period, prevents the restoration of ECM composition and re-establishment of pre-injury inflammation and wound healing gene expression levels. Regarding the mechanism of this effect, the analysis indicates that enhancement of expression of one or more of the genes described in Table 3 might be responsible. The most extreme activating effects of IBU on gene expression were for inflammatory mediators and ECM turnover components, including Cxcl5 (28-fold higher than without IBU at 25 d), Col3a1 (10-fold), Il6 (7.8-fold), Mmp9 (7.3-fold), Col5a1 (6.9-fold), Cxcl3 (6.1-fold) and Ptgs2 (5.9-fold).
At the histological level, the finding that IBU prevents the clearance of the HA inflammatory matrix indicates that it interferes with the phagocytic activity of tendon cells functioning as M2 tissue macrophages [47,48]. An expected downstream effect of persistent inflammatory activity at the 'wound site" would be the deposition of 'scar' collagens such as collagen types III and V, the expression of which was in fact found to be markedly activated by E-IBU dosing (Table 1). Although we were not able to detect extensive scar tissue deposition in the tendon body, either histologically or geometrically over the experimental time period used, it remains to be determined if the maintenance of IBU-treated tendinopathic mice, either at cage or treadmill activity [14], would generate pathological scarring in the tendon body and surrounding tissue. In the same context, 'angiogenic' stromal cell clusters in the peritenon and fat pad (Supplementary Figure 3) could progress to aberrant vascularization that has been reported in impaired wound healing [37] and in the stroma of rapidly growing tumors [49].
Whereas histologic analyses showed marked effects of IBU on tendon cell and ECM morphology at 25 d post-injury (Figure 4 and 5), biomechanical deficiencies were noted only for stiffness (p = 0.004) and elastic modulus (p = 0.012) for these tendons. This reflects sub-failure mechanical compromise caused here by IBU dosing both with and without TGF-β1 injury. In contrast, the absence of an effect of injury (with or without IBU) on maximum load, maximum stress and cross-sectional area shows that none of these treatments caused biomechanical changes typically associated with severe injury and rupture. Most notably, it was shown that IBU dosing of naïve mice for 22 days caused a 25-30% loss in stiffness and elastic modulus, which may be due to trended increases in displacement at maximum force (p = 0.09) and strain at maximum stress (p = 0.10) (Supplementary Figure 4). Taken together with increased expression of collagen types III, V, and IV, this data suggests the presence of an immature (un-crosslinked) and non-remodeled collagen matrix which contributes to larger strains, and decreased elastic modulus, similar to the use of glucocorticoids for tendon injury [50]. Further studies are warranted to confirm this phenomenon. In addition there was a consistent, general trend of increased spread (COV) in the E-IBU group for most parameters (Figure 6). We interpret this trend as biologically significant, consistent with the more marked effects of E-IBU than L-IBU on the gene expression profile (Table 3).
The current work supports the reported findings of deleterious effects of NSAIDs in the treatment of tendinopathies [11-13], but appears to conflict with the finding [51,52] that 7 days of oral IBU in patients with chronic Achilles tendinopathy produced no detectable changes in expression of COL1A1, TGF-β and PTGS2 in affected tendon tissue. The non-effect in the human studies however could be attributable to the short dosing period, chronic end-stage tendinopathy was studied and the dose (22.5 mg/kg/day) was low compared to the present model study (133 mg/kg/day). In addition, it may be relevant that a HA-HC-TSG6 inflammatory matrix and extensive scarring is found in the ligaments of DSLD affected horses [53] which are typically provided NSAIDs such as Phenylbutazone or Flunixin meglumine for pain relief (Brounts, S, personal communication).
To place the current findings in a broader clinical context, it appears useful to consider them in relation to the so-called continuum model [54,55], which predicts that treatment might be optimized (quote) "by tailoring interventions to the stage of pathology and targeting the primary driver (cell activation)". Indeed, the present study has provided support for the use of stage-specific treatments and has also generated novel information on the molecular aspects of the "cell activation" processes likely to be involved in different phases of the human pathology. The high animal to animal reproducibility of gene expression and histological changes in the model reported previously [3,14-16] and in the present study, revealed the existence of three phases, an initial inflammation (day 3), transition to a reparative phase (at day 9) and successful repair (by day 25) (see Results for details). In support of the importance of staged treatment, the start of IBU at day 3 resulted in poor repair at 25 days and over-expression of 50 target genes, whereas the start of IBU at 9 days enhanced repair and increased the expression of only 17 genes. Most notably, staged treatment also showed that early IBU dosing activated some genes unaffected by the TGFb1-injury itself and interestingly these have all been implicated in vascular remodeling (see Results for details), a feature of chronic tendinopathic tissue sections.
Our study with a non-surgically induced murine model of tendinopathy, showed that the use of Ibuprofen for pain relief during inflammatory phases of tendinopathy, might interfere with the normal processes of extracellular matrix remodeling and cellular control of expression of inflammatory and wound healing genes. In addition, the turnover of the post-injury inflammatory HA matrix was impaired, which may contribute to causing impaired cellular healing responses.
Although we were not able, using the current experimental design, to detect extensive scar tissue deposition in the tendon body or surrounding tissues, either histologically or biomechanically, it remains to be determined if more prolonged maintenance of IBU-treated tendinopathic mice, either at cage or treadmill activity would lead to such degeneration and eventual rupture of the tendon itself. Furthermore, to what extent the results obtained here can inform clinical decisions will require analysis of human post-injury tendons using the molecular probes we have described. If tissues from human tendon injury patients on anti-inflammatory medication become available, it will be interesting to determine whether the analgesic benefits are accompanied by unacceptable levels of pro-inflammatory matrix accumulation and pro-inflammatory macrophage polarization.
Funding was provided by the Rush Arthritis Institute (AB, AP), Katz/Rubschlager Endowment for OA Research (AP) and NIH (AR 63144) (VMW SR, JL, and KJS). We thanks Dr. Sabrina Brounts, U WI for helpful discussions regarding drug applications for pain management in DLSD affected horses.
National Institute of Health (NIAMS); Rush Arthritis Institute (RUMC).
Adam Bitterman: Study design, animal model experimentation, data analyses, manuscript preparation.
Shuguang Gao: Gene expression assays, data analyses.
Sabah Rezvani: Biomechanical testing, data analyses.
Jun Li: Animal model experimentation, histology methods.
Katie J Sikes: Animal model experimentation, gene expression data statistical analyses, manuscript preparation.
John Sandy: Study design, manuscript preparation.
Vincent Wang: Study design, manuscript preparation.
Simon Lee: Study design.
George Holmes: Study design.
Johnny Lin: Study design.
Anna Plaas: Study design, animal model experimentation, data analyses, immunohistochemical analyses, manuscript preparation.