
International Journal of Transplantation Research and Medicine
Blockade of ASC but not NLRP3 Inhibits DC Proliferation and T cell Activation in Response to Alloantigen
Sashi G Kasmisetty, Alana Shigeoka, Reza Elahimehr, Andrew Scheinok and Dianne B McKay*
Division of Nephrology and Hypertension, Department of Medicine, University of California San Diego, USA
*Corresponding author:
Dianne B McKay, MD, Division of Nephrology and Hypertension, Department of Medicine, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA, E-mail: d1mckay@ucsd.edu
Int J Transplant Res Med, IJTRM-2-016, (Volume 2, Issue 1), Research Article
Received: December 18, 2015: Accepted: January 13, 2016: Published: January 16, 2016
Citation: Kasmisetty SG, Shigeoka A, Elahimehr R, Scheinok A, McKay DB (2016) Blockade of ASC but not NLRP3 Inhibits DC Proliferation and T cell Activation in Response to Alloantigen. Int J Transplant Res Med 2:016
Copyright: © 2016 Kasmisetty SG, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
The NLRP3 inflammasome is a multimeric protein complex consisting of the sensor molecule NACHT, LRR and PYD domains-containing protein 3 (NLRP3), the adaptor molecule Apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and caspase-1. Molecules with conserved molecular motifs trigger the assembly of the inflammasome and lead to maturation of pro-inflammatory cytokines that induce a mature inflammatory response. Emerging data suggest that inflammasome activation in antigen presenting cells (APCs), such as dendritic cells (DCs), trigger their activation and ability to activate T cells. Our study asked whether blockade of the cytoplasmic NLRP3-inflammasome components NLRP3 and ASC interfered with alloantigen recognition and allograft rejection. We found that deletion of ASC significantly inhibited DC and T cell alloresponses, whereas deletion of NLRP3 had no effect. Despite the significant in vitro findings, there was no effect of the deletion of NLRP3 or ASC on allogeneic skin graft rejection across a stringent MHC barrier. The conclusion of this study is that while in vitro alloresponses were significantly inhibited by deletion of ASC, allograft rejection across a complete MHC mismatch was not prevented. The results suggest that the NLRP3-inflammasome coreceptor ASC plays a significant role by inhibiting allogeneic APC-induced T cell activation in vitro, independent of its inflammasome related effects.
Keywords
Inflammasome, NLRP3, ASC, Dendritic cells, Alloresponses, Allograft rejection
Introduction
Dendritic cells are professional APCs capable of stimulating a potent T cell response to alloantigen. Pro-inflammatory signals from innate immune pattern recognition receptors (PRRs) on donor DCs are thought to be key initiators of T cell responses [1-4]. The PRRs, such as toll-like receptors (TLRs) and nucleotide-binding oligomerization domain-like receptors (NLRs), have been recognized to be involved in some of the first, triggering, events leading to DC activation. TLRs have been extensively studied for their ability to mount an adaptive immune response, but less is known about the contribution of NLR family members, especially the inflammasome components in DC-induced allogeneic T cell responses [5-8].
Inflammasomes are cytoplasmic multiprotein complexes that mediate crucial innate immune signals during cellular damage. Danger signals during cellular damage trigger the assembly of one of the best studied inflammasomes, the NLRP3-inflammasome, comprised of NACHT, LRR and PYD domains-containing protein 3 (NLRP3), apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and pro-caspase-1. The activated NLRP3 inflammasome induces cleavage of pro-IL-1β and pro-IL-18 to produce mature IL-1β and IL-18. It has also been shown that inflammasome activation in DCs induces an IL1b-dependent adaptive immune response against tumors [9]. Other reports suggest ASC promotes inflammasome independent activity in DC maturation and antigen presentation [10]. Our study extended these studies to investigate whether blockade of the NLRP3-inflammasome components NLRP3 or ASC impacted DC function, alloantigen-induced T cell proliferation responses and/or allograft rejection.
Methods
Mice
All mice used in these experiments were housed in the vivarium at UCSD and approved for use by the Institutional Animal Care and Use Committee of the UCSD Animal Research Center. All animals were handled according to the recommendations of the Humanities and Sciences and the standards of the Association for Assessment and Accreditation of Laboratory Animal Care. BALB/cBYJ and C57BL/6J (WT) were obtained from Jackson Laboratories, Bar Harbor MN. The NLRP3- and ASC-deficient mice were a gift of H. Hoffman (UCSD). Caspase 1-deficient mice were obtained from R. Ulevitch (The Scripps Research Institute). All mice used in these experiments were bred onto a C57BL/6 background by more than 10 generations.
Dendritic cell function and T cell proliferation
Dendritic cells were isolated from spleens of either WT, NLRP3-deficient or ASC-deficient mice by positive selection using a CD11c+ MACS separation kit (R & D Systems, Minneapolis, MN). This purification method yields 90% purity of DCs in our hands. There were no differences in purity of CD11c DCs between the WT, NLRP3-deficient or ASC-deficient mice (data not shown). To test for proliferation, isolated DCs were plated on round-bottomed 96-well tissue culture dishes (Corning Costar, Cambridge, MA) and stimulated with LPS (1 μg/mL) or Pam (1 ug/mL) (Enzo Life Sciences, Inc., Farmingdale, NY) for 3 days. Proliferation was detected on serial days by MTT uptake (Promega, Madison, WI). Stimulated cells were stained with anti-CD86, anti-CD80 and anti-MHCII antibodies (BD Biosciences, San Jose, CA) to detect markers of activation. To test for migration, stimulated DCs were seeded at a density of 2 × 105 cells/well on a polycarbonate filter with 5 μm pore size in 24-well transwell chambers (Corning Costar, Cambridge, MA) in a total volume of 100 μL, incubated for 4 h at 37°C and detected by cell sorting using CellQuest software (Becton Dickinson, Franklin Lakes, NJ). The lower chambers contained 600 μL sterile 10K media alone (RPMI 1640 containing L-Glutamine (2 mM), FBS (10%), penicillin (100 U/ml), streptomycin (100 ug/ml), 2-ME (0.05 mM), HEPES (10 mM) or supplemented with 250 ng/mL CCL21 (R&D Systems, Minneapolis, MN) to induce migration. T cell stimulation (from BALB/cByJ mice) by WT, NLRP3-deficient or ASC-deficient derived DCs was detected using a standard mixed lymphocyte response (MLR), using methods previously published [11]. Allogeneic T cells were isolated from pooled peripheral LNs (axillary, brachial and inguinal nodes) from 3 BALB/cByJ mice per group. All mice were age and sex matched.
Allogeneic skin graft transplantation
Recipient Balb/cByJ mice (H-2d), or WT (C57BL/6), NLRP3-deficient or ASC-deficient mice (all H-2b) were anesthetized and the flank hair shaved with electric clippers. A graft bed was prepared on the lateral thoracic region under aseptic conditions. The graft bed was prepared by careful removal of the epidermis and dermis to the level of the panniculus carnosus, keeping the vascular bed undisrupted. Donor tail skin (from Balb/cByJ mice [H-2d], or WT, NLRP3-deficient or ASC-deficient mice (all H-2b)) was prepared by cutting the tail of a sacrificed donor mouse, incising circumferentially around the base of the tail and then down the dorsal surface and peeling off the donor skin. Equal-sized pieces were cut from the skin and kept in a wet sterile petri dish with PBS. The donor skin was then placed onto the vascular bed, leaving a margin of 1-2 mm on all sides. Syngeneic and allogeneic donor skin was placed into the same graft bed. The grafted skin was then covered with sterile, antibiotic (bacitracin)-impregnated Vaseline gauze, covered with a bandage and then wrapped in cloth tape. The grafts were left undisturbed for 7 days. On day 7 the bandages were removed and the grafts were photographed on a daily basis. Rejection was scored as 90% necrosis of the grafted tissue. Survival fractions were determined using the Kaplan-Meyer method. Comparison of survival curves and medial survival was performed using the log rank test provided by the Prism 4 software (GraphPad Software, La Jolla, CA).
Results
ASC exhibits inflammasome independent activities in antigen presenting cells (APCs)
Activation of the NLRP3 induced inflammasome in dendritic cells is known to activate adaptive T cell responses through a caspase-1 and IL-1b dependent pathway [9]. However, the role of the individual components of the inflammasome in a DCs ability to prime allogeneic T cells is not known. Therefore we asked whether the absence of either NLRP3 or ASC impacted the activation of DCs using an in vitro TLR2- and 4-induced DC activation model. We did not observe any differences in the proliferation of DCs (Figure 1A and Figure 1B), the expression of the activation markers MHCII, CD86 and CD80 (Figure 1D), or in the secretion of proinflammatory cytokine IFN-g (Figure 1C) in response to TLR2 (Pam 1 ug/mL) or TLR4 (LPS 1 ug/mL) ligands. Interestingly, Ippangunta et al. have shown that the inflammasome adaptor molecule ASC exhibits an inflammasome independent response in DCs through defective antigen presentation [10]. Therefore we hypothesized that absence of ASC in DCs might also induce defective alloantigen processing and presentation, thereby inhibiting an allogeneic T cell immune response. We investigated whether the WT, NLRP3-deficient and ASC-deficient (H-2b) DCs were able to induce proliferation of allogeneic Balb/cByJ (H-2d) T cells in a mixed lymphocyte reaction. We examined whether WT, NLRP3-deficient and ASC-deficient (H2b) T cells displayed defective proliferation in response to allogeneic Balb/cByJ (H2d) APCs in a reverse MLR. The results indicate that only the ASC deficiency in DCs significantly prevented them from stimulating T cell proliferation (Figure 1F). It was also observed that a deficiency in ASC, in T lymphocytes, rendered them inactive to antigen presentation from allogeneic APCs (Figure 1E). DC migration is a key step in the in vivo induction of an inflammatory response to transplanted tissue, so we also compared chemokine-induced migration between DCs from WT, NLRP3-deficient and ASC-deficient mice. Figure 1G shows that ASC-deficient DCs exhibit defective migration in response to the chemoattractant CCL21, suggesting a NLRP3-inflammasome independent activity for ASC in DCs.
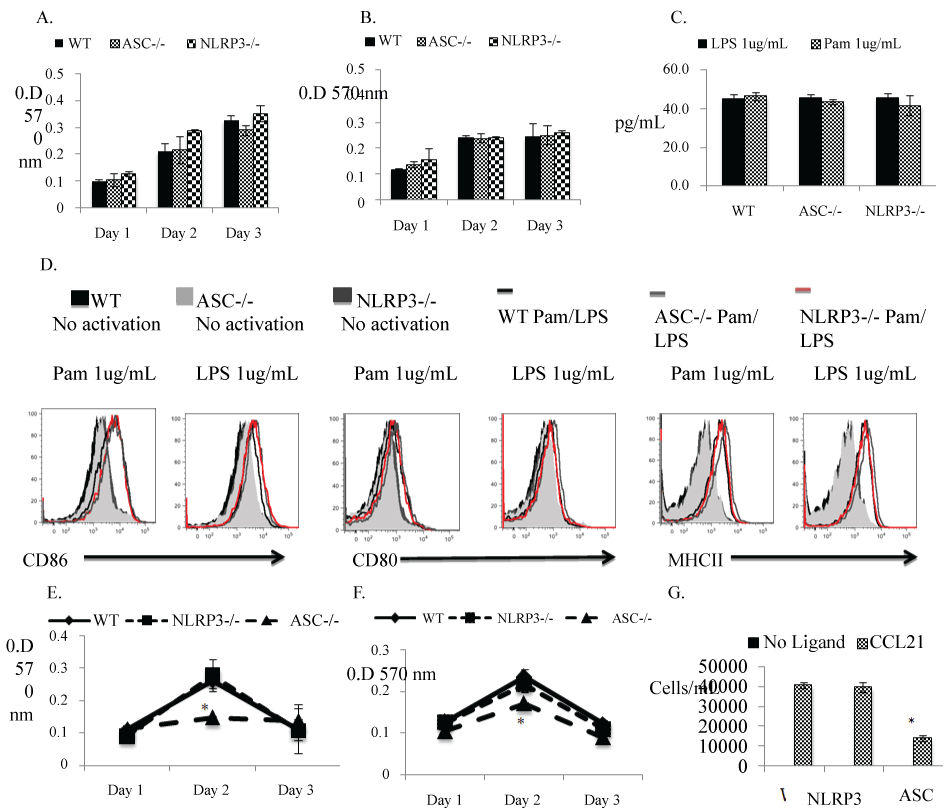
.
Figure 1: Absence of ASC in DCs significantly affects their ability to induce allogeneic T cell proliferation:
(A) DCs isolated from spleens of WT vs. ASC-deficient vs. NLRP3-deficient mice were stimulated with LPS (1 μg/ml) for 3 consecutive days. Proliferation was detected by MTT uptake by measuring optical density at 570 nm. The bargraphs represent increase in proliferation compared to unstimulated DCs. Error bars represent SD of three samples. The graph represents one of three identical experiments; (B) DCs isolated from spleens of WT vs. ASC-deficient vs. NLRP3-deficient mice were stimulated with Pam (1 μg/ml) for 3 consecutive days. Proliferation was detected by MTT uptake by measuring optical density at 570 nm. The bargraphs represent increase in proliferation compared to unstimulated DCs. Error bars represent SD of three samples. The graph represents one of three identical experiments. Error bars represent SD of three samples. The graph represents one of three identical experiments; (C) Increase in IFN-g secretion (pg/mL) detected in supernatants of MLR cultures from WT vs. ASC-deficient vs. NLRP3-deficient DCs stimulated with LPS (1 μg/ml) or Pam (1 ug/mL) for 24 hr compared to unstimulated DCs; (D) MHCII, CD86 and CD80 expression on WT vs. ASC-deficient vs. NLRP3-deficient dendritic cells stimulated with LPS (1 μg/ml) or Pam (1 ug/mL) for 24 hr, and stained with MHCII, CD86 and CD80 antibodies and analyzed by FACS. This represents one of 3 identical experiments; (E) Balb/c irradiated DCs (H-2d) cultured with allogeneic WT vs. ASC-deficient vs. NLRP3-deficientT cells (H-2b) in a standard MLR assay. Proliferation was detected by 3H-thy uptake on the 4th day of culture. Error bars represent SDs; graph represents 1 of 3 identical experiments; (F). WT vs. ASC-deficient vs. NLRP3-deficient irradiated DCs (all H-2b) cultured with allogeneic T cells (H-2d) in MLR assay. Proliferation was detected by 3H-thy uptake on the 4th day of culture. Error bars represent SDs; graph represents 1 of 3 identical experiments; (G) DC migration was detected in a transwell assay comparing WT vs. ASC-deficient vs. NLRP3-deficient DCs stimulated with LPS (1 ug/mL) or Pam (1 ug/mL) overnite and allowed to migrate for 4 h in response of chemokine CCL21 (250 ng/mL) in a transwell chamber. Error bars represent SD of three samples. The graph represents one of three identical experiments.
*represents significance value p < 0.05.
View Figure 1
Allogeneic skin grafts lacking both NLRP3 and ASC are not protected from rejection
Donor APCs play an important role in vivo by initiating host responses to transplanted solid organ allografts. Following skin transplantation, sentinel DCs that reside in the transplanted donor tissue are activated, leading to their subsequent migration to host draining lymph nodes where host T cells are activated [12]. Blockade of DC migration from the transplanted skin can prevent rejection of transplanted allografts in experimental models [13,14]. Since the absence of ASC affected the ability of DCs to effectively stimulate allogeneic T cells in vitro and to migrate in response to chemokine, we next asked whether the absence of ASC or NLRP3 proteins in donor DCs (donor skin from ASC-deficient and NLRP3-deficient mice) would impact the rejection of skin grafts on allogeneic hosts. As shown in figures 2B and Figure 2D, the absence of ASC or NLRP3 in the donor skin did not affect the kinetics of skin graft rejection (Figure 2D). A representative picture of the skin grafts taken on day 13 is shown in figure 2B, with a syngeneic control graft shown on the right of the allogeneic grafts on the same recipient. Shown in figure 2A and figure 2C, the absence of ASC or NLRP3 in the recipient had no effect on the rate of allogeneic skin graft rejection, suggesting that indirect antigen presentation also was not impacted by ASC deficiency.
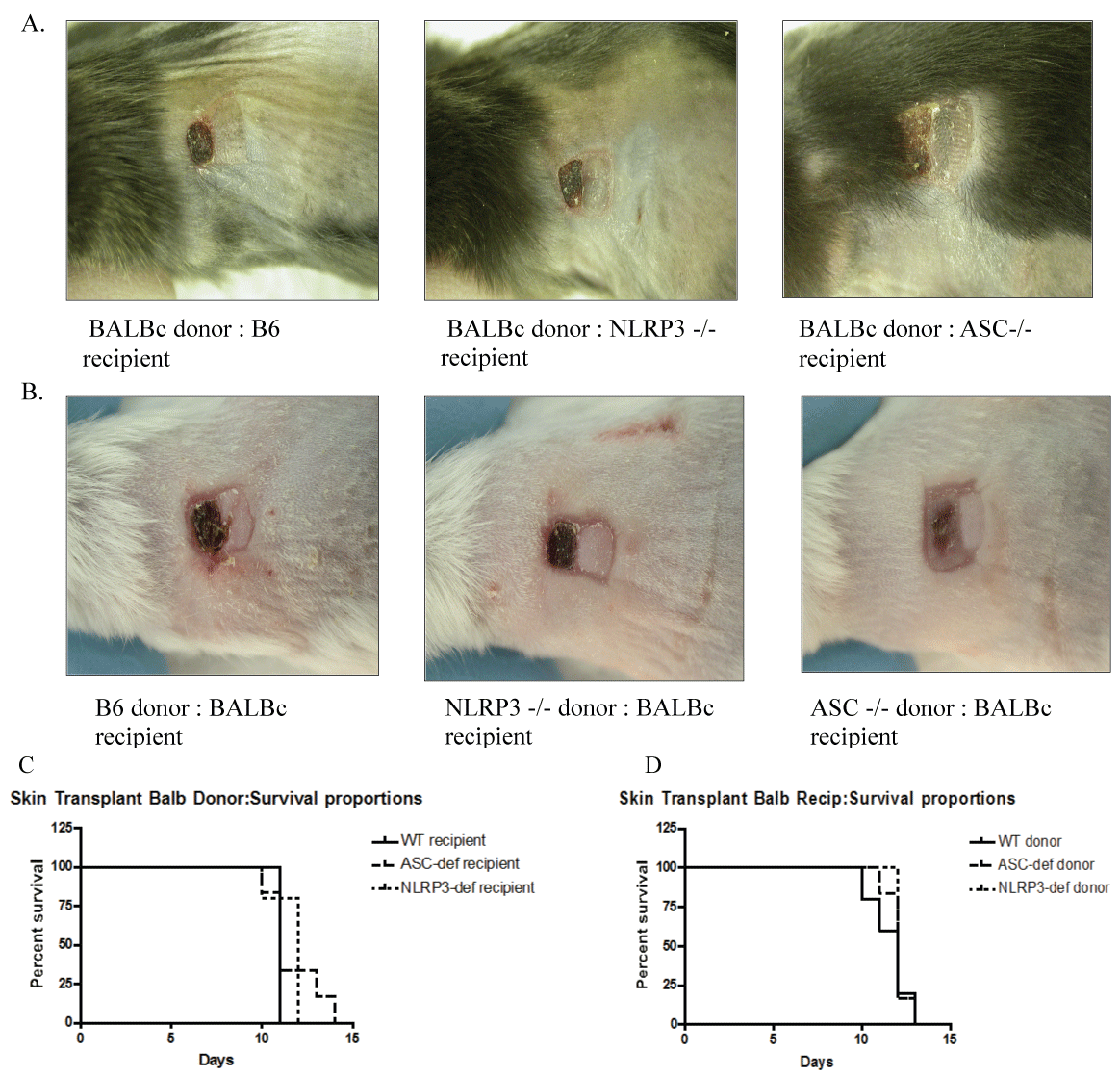
.
Figure 2: Skin graft rejection in WT recipients of NOD-deficient recipients. (A & B) Representative photograph of syngeneic (right) and allogeneic (left) skin grafts (H-2d) on recipient micenoted by title under each photograph. Representative photograph taken on day 13; (C) Percent survival of donor skin grafts (Balb/c, BYJ H-2d) on allogeneic recipients (all H-2b), noted by donor in legend, n = 6 mice/gp; (D) Percent survival of donor skin grafts (H-2b) in allogeneic recipients (BALB/cByJ, H-2d), n = 6 mice/gp.
View Figure 2
Discussion
Inflammasome activation in DCs has been shown to induce T cell priming both in vitro and in vivo, and therefore we hypothesized that the NLRP3 inflammasome might play an important role in adaptive immune responses to alloantigen. This study investigated the role of NLRP3 and its adaptor molecule ASC in DC activation and alloantigen presentation and asked whether defects in either inflammasome component impacted in vitro and in vivo alloresponses.
Our first studies asked whether the absence of NLRP3 or ASC modified DC activation and function. We found that the absence of either protein had no impact on TLR2- and TLR4-induced DC proliferation, or expression of activation markers; suggesting that NLRP3 and ASC were not critical components for DC activation in our model.
Several studies have shown the importance of the NLRP3 inflammasome in DC responses to alloantigen. The absence of NLRP3 protects from graft versus host disease (GVHD) after allogeneic stem cell transplants [15] and activation of the NLRP3 inflammasome has been associated with a DC induced anti- T cell response tumor responses [9], and the absence of ASC inhibits T cell priming and protects from collagen induced arthritis, an effect which appears to be independent of the NLRP3-inflammasome [16]. Interestingly, our study showed that a deficiency in NLRP3 in DCs had no impact on allogeneic T cell proliferative responses, whereas in contrast the absence of ASC in DCs was associated with a significant decrease in activation of allogeneic T cells. We also found that the absence of ASC had a significant impact on chemokine-induced DC migration. NLRP3 deficient DCs exhibited no difference in their migratory ability from WT DCs. Thus we identified that ASC has an NLRP3-inflammasome independent function in DCs, and plays a key role in DC migration and priming of allogeneic T cells.
Others have reported that NLRP3 exerts an inflammasome independent transcriptional response in CD4+ T cells, whereas ASC fails to show similar activity in T cells [17]. Focusing on the role of these components in T cells, we also found that there was discordance between the NLRP3 inflammasome components. T cells deficient in ASC have significantly impaired proliferative responses to alloantigen, whereas a T cell deficiency in NLRP3 has no impact on alloantigen-induced T cell proliferation.
Rejection of donor skin grafts involves migration of “passenger donor dendritic cells” from the transplanted graft to host lymph nodes, where activation of naïve host T lymphocytes initiates the cascade of graft rejection [18]. Since we noted that the absence of ASC in both DCs and T cells impacted in vitro alloresponses, we next tested in vivo alloresponses using a skin graft model across a stringent MHC barrier (H-2b to H-2d). Donor skin deficient in NLRP3 and ASC versus skin from WT mice (all H2-b) was transplanted onto allogeneic hosts (H2-d). Interestingly, even though blockade of ASC significantly impacted T cell alloreactivity in vitro, there was no effect noted in vitro as measured by the rejection of the ASC-deficient skin grafts. Furthermore, there were no differences in rejection kinetics noted when the reverse transplants were performed – the skin from BALB/c mice (H-2d) transplanted onto NLRP3-deficient or ASC-deficient hosts showed the same rejection kinetics as donor WT skin from C57BL/6 mice. Our data demonstrate that the absence of ASC confers an NLRP3-independent defect on DC activation, migration and allopresentation; and T cells deficient in ASC also have significant proliferative defects. Interestingly, in vivo alloresponses were not affected by the deficiency in ASC, suggesting that additional investigations need to be tested across a less stringent allogeneic barrier. These studies are currently ongoing in our laboratory, as well as studies to better understand how ASC impacts DC and T cell responses to alloantigen.
Acknowledgements
The work was supported by NIH grants (R01 DK091136, R01 DK075718) and California Institute of Regenerative Medicine grant (CIRM RB5-07379).
References
-
Rigoryev DN, Liu M, Hassoun HT, Cheadle C, Barnes KC, et al. (2008) The Local and Systemic Inflammatory Transcriptome after Acute Kidney Injury. J Am Soc Nephrol 19: 547-558
-
Tsan MF, Gao B (2004) Endogenous ligands of Toll-like receptors. J Leukoc Biol 76: 514-519.
-
Jang HR, Rabb H (2009) The innate immune response in ischemic acute kidney injury. Clin Immunol 130: 41-50.
-
Cook DN, Pisetsky DS, Schwartz DA (2004) Toll-like receptors in the pathogenesis of human disease. Nat Immunol 5: 975-979.
-
Ben Mkaddem S, Chassin C, Vandewalle A (2010) Contribution of renal tubule epithelial cells in the innate immune response during renal bacterial infections and ischemia-reperfusion injury. Chang Gung Med J 33: 225-240.
-
Bolisetty S, Agarwal A (2009) Neutrophils in acute kidney injury: not neutral any more. Kidney Int 75: 674-676.
-
Iwasaki A, Medzhitov R (2004) Toll-like receptor control of the adaptive immune responses. Nat Immunol 5: 987-995.
-
Schnare M, Barton GM, Holt AC, Takeda A, Akira S, et al. (2001) Toll-like receptors control activation of adaptive immune responses. Nat Immunol 2: 947-950.
-
Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, et al. (2009) Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β-dependent adaptive immunity against tumors. Nat Med 15: 1170-1178.
-
Ippagunta SK, Malireddi RKS, Shaw PJ, Neale GA, Walle LV, et al. (2011) The inflammasome adaptor ASC regulates the function of adaptive immune cells by controlling Dock2-mediated Rac activation and actin polymerization. Nat Immunol 12: 1010-1016.
-
McKay D, Shigeoka A, Rubinstein M, Surh C, Sprent J (2006) Simultaneous deletion of MyD88 and Trif delays major histocompatibility and minor antigen mismatch allograft rejection. Eur J Immunol 36: 1994-2002.
-
Peiser M, Koeck J, Kirschning CJ, Wittig B, Wanner R (2008) Human Langerhans cells selectively activated via Toll-like receptor 2 agonists acquire migratory and CD4+ T cell stimulatory capacity. J Leukoc Biol 83: 1118-1127.
-
Barker CF, Billingham RE (1968) The role of afferent lymphatics in the rejection of skin homografts. J Exp Med 128: 197-221.
-
Lakkis FG, Arakelov A, Konieczny BT, Inoue Y (2000) Immunologic 'ignorance' of vascularized organ transplants in the absence of secondary lymphoid tissue. Nat Med 6: 686-688.
-
Jankovic D, Ganesan J, Bscheider M, Stickel N, Weber FC, et al. (2013) The Nlrp3 inflammasome regulates acute graft-versus-host disease. JEM 210: 1899-1910.
-
Ippagunta SK, Brand DD, Luo J, Boyd KL, Calabrese C, et al. (2010) Inflammasome-independent Role of Apoptosis-associated Speck-like Protein Containing a CARD (ASC) in T Cell Priming Is Critical for Collagen-induced Arthritis. J Biol Chem 285: 12454-12462.
-
Bruchard M, Rebé C, Derangère V, Togbé D, Ryffel B, et al. (2015) The receptor NLRP3 is a transcriptional regulator of TH2 differentiation. Nat Immunol 16: 859-870.
-
Larsen CP, Steinman RM, Witmer-Pack M, Hankins DF, Morris PJ, et al. (1990) Migration and maturation of Langerhans cells in skin transplants and explants. J Exp Med 172: 1483-1493.
