

Journal of Geriatric Medicine and Gerontology
Phase II Clinical Trial of Nicotinamide for the Treatment of Mild to Moderate Alzheimer's Disease
Michael J Phelan1,2, Ruth A Mulnard3, Daniel L Gillen1,2 and Steven S Schreiber2,4,5*
1Department of Statistics, University of California, USA
2Institute for Memory Impairments and Neurological Disorders, University of California, USA
3Program in Nursing, University of California, USA
4Departments of Neurology, Anatomy & Neurobiology, Psychiatry & Human Behavior, University of California, USA
5Neurology Section, VA Long Beach Healthcare System, USA
*Corresponding author: Steven S Schreiber, Departments of Neurology, Anatomy & Neurobiology, Psychiatry & Human Behavior, University of California, Irvine, School of Medicine, 106 Irvine Hall, Irvine, CA 92697-4275, USA, Tel: 949-824-6402, E-mail: sschreib@uci.edu
J Geriatr Med Gerontol, JGMG-3-021, (Volume 3, Issue 1), Research Article; ISSN: 2469-5858
Received: July 26, 2016 | Accepted: February 10, 2017 | Published: February 14, 2017
Citation: Phelan MJ, Mulnard RA, Gillen DL, Schreiber SS (2017) Phase II Clinical Trial of Nicotinamide for the Treatment of Mild to Moderate Alzheimer's Disease. J Geriatr Med Gerontol 3:021. 10.23937/2469-5858/1510021
Copyright: © 2017 Phelan MJ, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Background: Disease-modifying treatments for Alzheimer's disease (AD) are currently unavailable and are the focus of an intensive research effort. We found vitamin B3, nicotinamide (NA), to significantly reduce pathology and improve behavior in AD transgenic mice. These results led us to conduct a double-blind, placebo-controlled randomized clinical trial of NA in mild to moderate AD.
Methods: Following randomization, subjects received either NA (n = 15, 1500 mg twice daily) or placebo (n = 16) for 24 weeks. A battery of outcome measures were obtained at baseline and 6- week intervals and included the AD Assessment Scale-Cognitive Subscale, Clinician's Interview-Based Impression of Change Plus Caregiver Input, AD Cooperative Study-Activities of Daily Living Scale, and Clinical Dementia Rating Scale.
Results: There were no significant effects of NA on the primary or secondary endpoints. A mild effect of low compliance was observed on word recall and command tasks. There were no differences in adverse events experienced by NA- and placebo-treated groups.
Conclusions: This study failed to demonstrate that extended-release NA, or vitamin B3, improves cognitive function in subjects with mild to moderate AD over 24 weeks. The lack of efficacy of NA may have been due to several contributing factors including a low sample size, inclusion of subjects with moderate AD, and a relatively short treatment phase. The results also show that high dose NA is relatively safe in elderly subjects with AD. With the current emphasis on the early diagnosis and treatment of AD, a longer duration of treatment with NA in subjects with preclinical AD and/or mild cognitive impairment (MCI) may be warranted.
Keywords
Nutraceutical, Vitamin, Alzheimer's disease, Cognitive function
Introduction
Alzheimer's disease (AD) is the leading cause of age-related dementia in the elderly [1]. Progressive cognitive decline due to AD is associated with the accumulation, in selected brain regions, of beta-amyloid and hyperphosphorylated tau into amyloid plaques and neurofibrillary tangles, respectively [1]. Over the years these major pathological features of AD have become the primary therapeutic targets of drug development strategies. One group of drugs that has generated enormous interest for the potential treatment of AD is naturally-occurring compounds or nutraceuticals, which includes vitamins and other nutritional supplements [2]. In particular, supplementation with vitamin E and B vitamins, i.e., thiamine, folic acid, B6, and B12, has recently been explored in AD clinical trials. The results of studies with vitamins and other nutraceuticals have thus far been modest [2,3].
Nicotinamide (NA) or niacinamide, is the water soluble amide form of niacin (vitamin B3) [4]. NA is a key component of nicotinamide adenine dinucleotide (NAD), a co-enzyme involved in many cellular oxidation-reduction reactions [4]. In addition to its role as a co-factor, NA has been shown to act as a free radical scavenger, and modulator of both immune cell function and apoptosis [4]. Importantly, NA is an inhibitor of the class III histone deacetylases (HDACs), or sirtuins [4,5]. Sirtuins, particularly Sirt1, are involved in many key cellular functions and have been implicated in aging [6,7]. NA was first isolated in 1935 and has been employed in clinical trials for a variety of disorders over the last four decades [8-10]. Studies have shown that NA is relatively safe at doses of up to several grams per day [10]. The pharmacokinetics of NA depends on dose, species, gender, and route of administration [11]. NA is readily absorbed from the gastrointestinal tract [12]. Peak serum concentrations are reached in humans within one hour of oral ingestion of standard preparations [13].
In AD triple transgenic mice, NA selectively decreased the accumulation of phosphorylated tau in vulnerable brain regions and significantly improved cognitive decline, without affecting the accumulation of beta-amyloid [14]. In this study, NA caused a marked increase in the levels of acetylated α-tubulin and microtubule-associated protein 2c (MAP2c), both of which promote microtubule stabilization. The effects of NA on tau pathology were also reproduced by genetic reduction of Sirt1 levels [14]. Given the finding of selective targeting of tau pathology in an animal model of AD and the benign safety profile of NA in non-AD clinical trials, we performed a randomized double-blind placebo-controlled study to test the safety and efficacy of NA in subjects with mild to moderate AD.
Materials and Methods
The study was conducted in compliance with guidelines on human experimentation under protocols approved by the Institutional Review Boards of the University of California, Irvine, School of Medicine and the VA Long Beach Healthcare System (VALBHS). Subjects were recruited through the UC Irvine Alzheimer's Disease Research Center (ADRC) and the outpatient Neurology clinics of the VALBHS. Inclusion criteria for the study included a minimum age of 50 years, a diagnosis of mild to moderate dementia based on a Mini-Mental State Examination [MMSE] score between 13 and 25, brain imaging (computed tomographic scan or magnetic resonance image) consistent with a diagnosis of probable AD based on published criteria [15], Hachinski Ischemic Score < 4, maintenance of stable dosing of cholinesterase inhibitors (ChEIs) and/or memantine for at least 30 days, and a caregiver/relative available who could assist with supplement administration and accompany the subject to all study visits. Subjects were excluded from the study if they were diagnosed with dementia due to another cause, had other neurological or psychiatric diseases including pseudodementia, an unstable medical condition, a history of alcoholism, drug abuse, liver disease or peptic ulcer disease, started a ChEI, memantine or any investigational drug within 30 days of screening, were taking a supplement containing NA, were pregnant or had the potential to become pregnant.
This was a randomized, double-blind, placebo-controlled study. Following informed consent, a screening evaluation was performed that included measurement of vital signs, physical and neurological examinations, MMSE-score, and a blood draw for complete blood count (CBC), serum electrolytes and liver function tests (LFTs). Within 1-2 weeks following screening subjects that met the inclusion criteria were randomly assigned to receive either extended release Nicotinamide [NA] (Endur-amide [Niacinamide, vitamin B3] 1500 mg orally twice a day (Endurance Products Company, Tigard, OR), or a placebo identical in size, shape and color to NA, for 24 weeks. A one-to-one randomization scheme generated via a computerized random number generator was used to assign participants to either the treatment or placebo group. Preparations were dispensed in numerically coded bottles. The allocation sequence was concealed from participants and all members of the research team for the entire duration of the study. The dose of NA was equivalent to that used in other clinical trials and approximated the dose used in our pre-clinical study. A baseline evaluation included completion of the cognitive subscale of the Alzheimer's disease Assessment Scale (ADAS-cog), Clinician's Interview-Based Impression of Change Plus Caregiver Input (CIBIC-Plus), Alzheimer's Disease Cooperative Study-Activities of Daily Living Scale (ADCS-ADL), and Clinical Dementia Rating Scale (CDR). An early safety visit was conducted at week 4 to check LFTs, serum electrolytes and CBC. The ADAS-cog, CIBIC-Plus, ADCS-ADL and CDR were again completed at weeks 6, 12, 18 and 24. At week 24 vital signs, physical and neurological examinations, and MMSE were also completed. Standard pill counts, study drug compliance and adverse events were recorded at each visit.
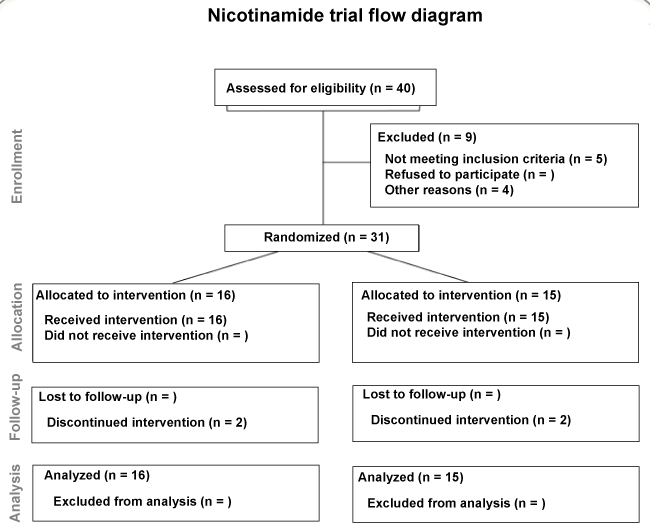
The primary outcome measure for the trial was total performance on the ADAS-cog. Secondary outcome measures included performance on the CIBIC-Plus, ADCS-ADL and CDR. To address the a priori hypothesis that use of NA would improve mean cognitive function in subjects with mild to moderate Alzheimer's disease, the primary analysis tested the effect of treatment on the mean 24-week change from baseline in ADAS-cog. An analysis of covariance (ANCOVA) was used to estimate treatment effects on within-subject change in mean ADAS-cog score over 24 weeks [16]. Specifically, the 24-week ADAS-cog score was regressed on an indicator of treatment and baseline ADAS-cog. In this case, a test of the coefficient for the treatment indicator equaling zero is equivalent to a test of the treatment effect. All secondary endpoints were analyzed using this approach. Holm's method was used for significance testing of secondary endpoints to adjust p-values for multiple comparisons [17]. Adverse events were described qualitatively. The trial was designed to attain 80% power when the true mean difference between treatment and control was one standard deviation. The planned sample size was for 25 participants per treatment group. Ultimately, 40 subjects were enrolled and 31 subjects randomized (Figure 1). Demographic data (continuous variables) were analyzed by two sample t-test.
Results
A Consort flow diagram in which the number of subjects enrolled, randomized and assessed in the study is shown in Figure 1. Thirty-one participants (100%) remained in the study through visits 2, 4 and 5, twenty-nine participants (94%) remained through visit 6, and twenty-eight (90%) through visit 7. Two subjects in each group withdrew from the study due to progression of illness or moving out of the area. The demographic characteristics of the study population are presented in Table 1. The average age of the study participants was 79 years in both groups. As shown in Table 1, the groups were comparable with respect to ethnicity, education, weight, height, MMSE score and body mass index (BMI). Fifty-six percent of subjects in the treatment group were male, compared to 80% randomized to placebo. Average baseline MMSE-score was 22 points in both groups. From pill counts, mean treatment compliance throughout the study was 72% in both the treatment and control groups.
![]()
Table 1: Baseline characteristics of the subjects by treatment. Continuous variables are reported as mean ± standard deviation, and categorical variables are reported as counts and proportions.
View Table 1
We first examined whether there was any significant change in cognitive function within each group over the course of the treatment period. The estimated mean within-subject change in the primary endpoint, ADAS-cog, from baseline to week 24 in the placebo group was -0.067 points (estimate = -0.067, 95% CI -2.78, 2.64, p-value = 0.96). In the group that received NA, the mean within-subject change from baseline to week 24 was -0.42 points (estimate = -0.41, 95% CI -2.72, 1.89, p-value = 0.70).
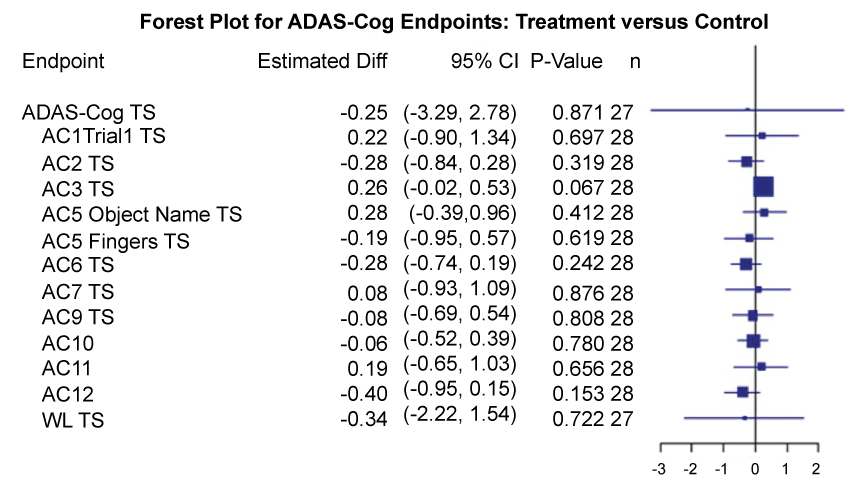
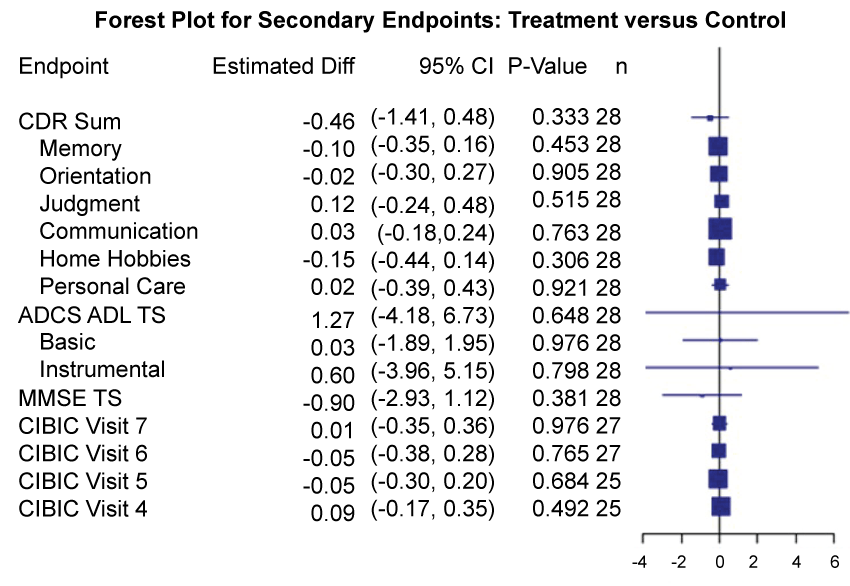
The results for the primary and secondary endpoints between treatment groups are presented in the forest plots in Figure 2 and Figure 3, respectively. These figures show the estimated mean difference in each endpoint and corresponding 95% confidence intervals (unadjusted for multiple comparisons). No effect of NA was observed for the primary or any of the secondary endpoints. Specifically, within subject change in mean ADAS-cog over 24 weeks was estimated to be -0.25 points lower in the treatment arm when compared to the placebo arm (95% CI: -3.29, 2.78; p = 0.88). Similarly, the estimated effects of treatment on all secondary endpoints at 24 weeks were not significant; for example, the estimated effect on total score for CDR Sum was -0.46 points (95% CI: -1.41, 0.48; p = 0.33).
.
Figure 2: Forest plots for treatment comparisons.
Forest plot of treatment effects on ADAS-Cog endpoints, including estimated mean difference between treatment and control, 95% confidence interval (CI), p-values, and number of complete cases (n). P-values and confidence intervals are unadjusted for multiple comparisons. Boxes plotted at mean treatment differences are drawn proportional to the standard error. The unadjusted 95% CI for each co-primary endpoint covered zero.
View Figure 2
.
Figure 3: Forest plots for treatment comparisons.
Forest plot of treatment effects on secondary endpoints, including estimated mean difference between treatment and control, 95% confidence interval (CI), p-values, and number of complete cases (n). P-values and confidence intervals are unadjusted for multiple comparisons. Boxes plotted at mean treatment differences are drawn proportional to the standard error. The unadjusted 95% CI for each secondary endpoint covered zero.
View Figure 3
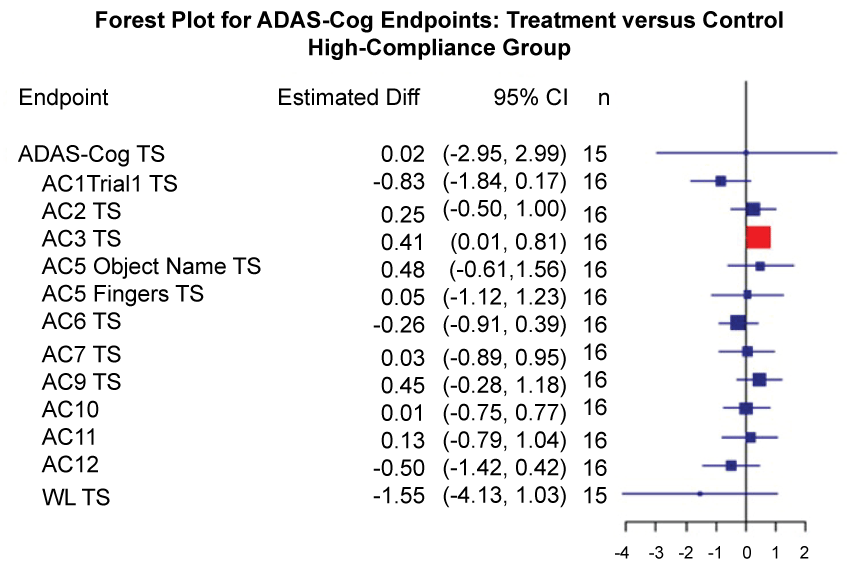
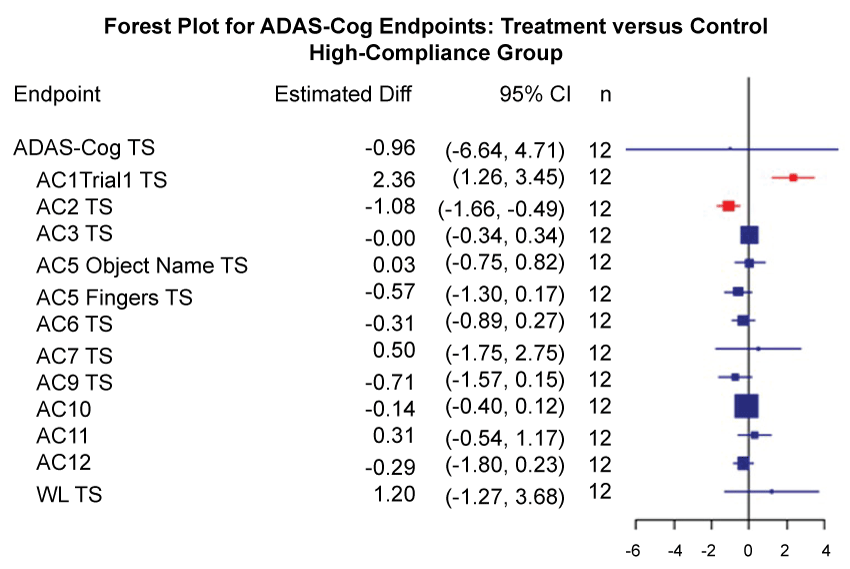
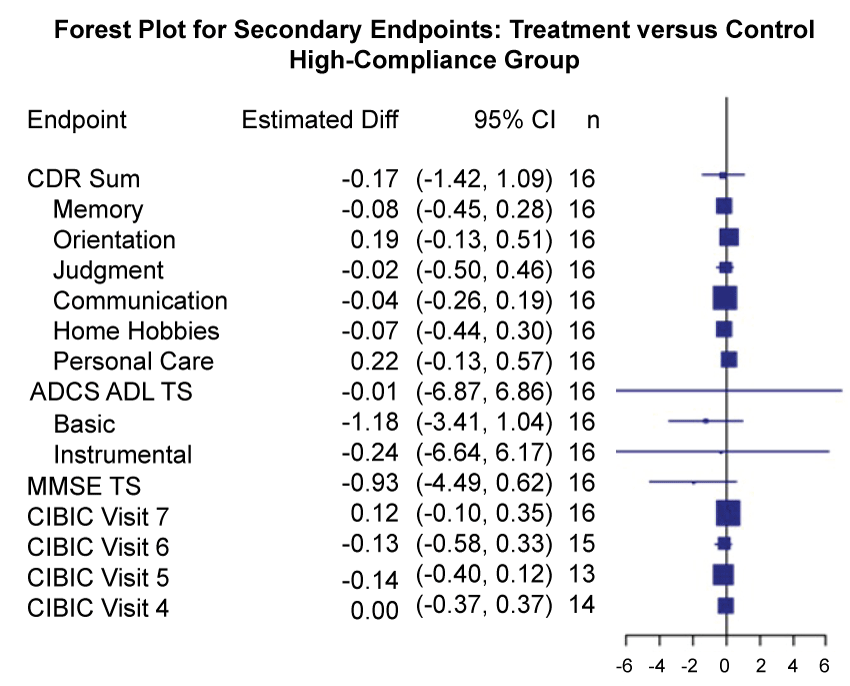
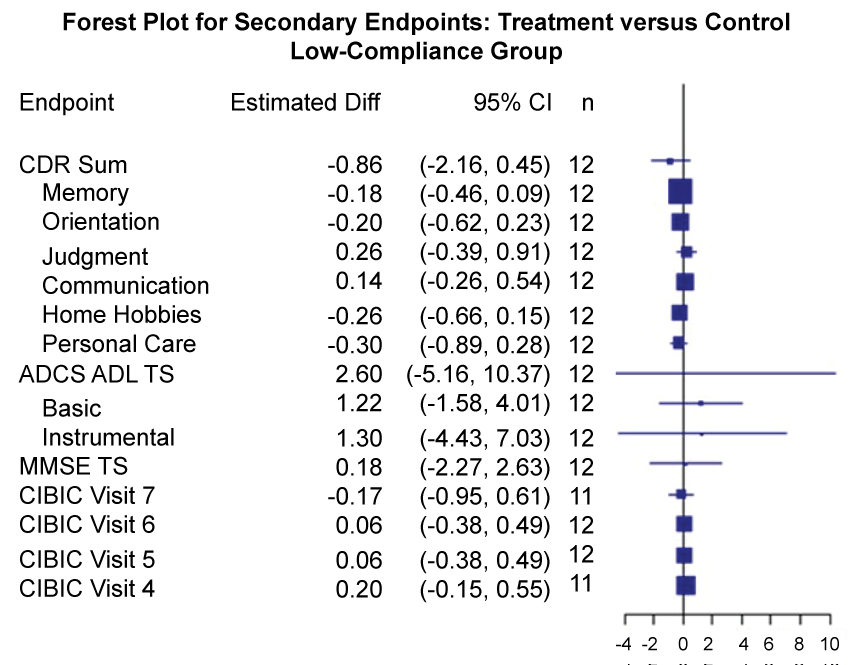
To investigate the role of compliance on the outcomes of the trial, the participants were divided at the median compliance rate (above 72% compliant and below 72% compliant). The results shown in Figure 4, Figure 5, Figure 6 and Figure 7 report the estimated mean treatment difference and unadjusted 95% confidence interval for each endpoint. For the high compliance group, the estimated effect of treatment on mean change in ADAS-cog over 24 weeks was 0.02 (95% CI: -2.95, 2.99; p = 0.99, Figure 4). Similarly, for the low compliance group, the estimated effect was -0.96 (95% CI:-6.44, 4.71; p = 0.74, Figure 5). On average, high-versus-low compliance was associated with +1.6 (95% CI: -1.66, 4.80) point difference in the 24-week change from baseline in ADAS-cog; the difference was not significant (p = 0.34). All unadjusted confidence intervals included zero with two exceptions: among low-compliance participants the ADAS-Cog subcomponent AC1 Trial1 TS (word recall) was somewhat higher in the treatment group, i.e., worse performance, but the AC2 TS subcomponent (commands) was slightly lower (Figure 5). The corresponding p-values were 0.00001 for AC1 Trial1 TS and 0.0003 for AC2 TS, which remained statistically significant after implementing the Holm's adjustment for multiple comparisons. The role of compliance was explored in post-hoc comparisons of treatment vs. control groups for all secondary endpoints (Figure 6 and Figure 7). There was no effect of compliance on secondary outcome measures.
.
Figure 4: Forest plots for treatment comparisons by compliance (Primary Outcome Measures).
Forest plot of treatment effects on ADAS-Cog endpoints among the high-compliance group, including estimated mean difference between treatment and control, 95% confidence interval (CI), p-values, and number of complete cases (n). P-values and confidence intervals are unadjusted for multiple comparisons. Boxes plotted at mean treatment differences are drawn proportional to the standard error. The red color denotes a confidence interval that excluded zero. The unadjusted 95% CI for each co-primary endpoint covered zero, with the exception of AC3TS.
View Figure 4
.
Figure 5: Forest plots for treatment comparisons by compliance (Primary Outcome Measures).
Forest plot of treatment effects on ADAS-Cog endpoints among the low-compliance group, including estimated mean difference between treatment and control, 95% confidence interval (CI), p-values, and number of complete cases (n). P-values and confidence intervals are unadjusted for multiple comparisons. Boxes plotted at mean treatment differences are drawn proportional to the standard error. The red color denotes a confidence interval that excluded zero. The unadjusted 95% CI for each co-primary endpoint covered zero, with the exception of AC1 Trail1 TS and AC2 TS.
View Figure 5
.
Figure 6: Forest plots for treatment comparisons by compliance (Secondary Outcome Measures).
Forest plot of treatment effects on secondary endpoints among the high-compliance group, including estimated mean difference between treatment and control, 95% confidence interval (CI), p-values, and number of complete cases (n). P-values and confidence intervals are unadjusted for multiple comparisons. Boxes plotted at mean treatment differences are drawn proportional to the standard error. The unadjusted 95% CI for each secondary endpoint covered zero.
View Figure 6
.
Figure 7: Forest plots for treatment comparisons by compliance (Secondary Outcome Measures).
Forest plot of treatment effects on secondary endpoints among the low-compliance group, including estimated mean difference between treatment and control, 95% confidence interval (CI), p-values, and number of complete cases (n). P-values and confidence intervals are unadjusted for multiple comparisons. Boxes plotted at mean treatment differences are drawn proportional to the standard error. The unadjusted 95% CI for each secondary endpoint covered zero.
View Figure 7
Adverse events in the form of very brief non-threatening visual hallucinations or delusions were reported by 4 subjects receiving NA and 4 subjects receiving placebo. No adverse event resulted in subject withdrawal from the study. At 4 weeks, there were no adverse effects of NA on serum electrolytes, CBC or LFTs.
Discussion
The results of this study fail to demonstrate that extended-release NA, or vitamin B3, improves cognitive function in subjects with mild to moderate AD over 24 weeks. Specifically, the primary outcome (ADAS-Cog TS) and secondary outcome measures in subjects taking NA were not significantly different from those taking placebo. One of the major challenges in clinical research that is particularly relevant to Alzheimer's clinical trials is treatment compliance [18]. In the present study the mean compliance rate was 72% for both the treatment and control groups. Analyses of low and high compliance groups revealed a slight effect of low compliance on the word recall and commands subscales of the ADAS-Cog. Although these findings may merit follow-up in future research, in the present study compliance appeared to play a negligible role in the outcomes of this trial.
In addition to its function as an HDAC inhibitor there are other functions of NA that make it an attractive neurotherapeutic [4,5]. Accordingly, additional reports showed robust effects of NA in animal models of AD as well as other neurodegenerative diseases [19-24]. However, while NA and other nutraceuticals have shown promise in preclinical studies, a comparable level of success in AD clinical trials has yet to be achieved [2]. In the present study several factors may have contributed to the lack of efficacy of NA. First, the proposed sample size (25 in each group) was not attained, and the total number of subjects enrolled in each group was likely too low to detect a mean difference of one standard deviation. Further, the 24 week duration of treatment may have precluded the observance of any long-term effects of NA. Finally, the inclusion of subjects with moderate AD may have also negatively impacted the results. Drawing a parallel with our preclinical study, NA did not significantly improve cognition in one year old transgenic mice with more advanced AD-related pathology [14]. Importantly, we conclude that high dose NA is relatively safe in elderly subjects with AD. Thus, with the current emphasis on the early diagnosis and treatment of AD, it may be worthwhile to investigate whether NA may be beneficial in patients with preclinical AD and/or MCI.
Acknowledgements
This study was supported by a grant from the Alzheimer's Association to SSS. The authors wish to thank Catherine McAdams-Ortiz, Deeba Sultani, Beatriz Yanez, Drs. Gaby Thai and Arnold Starr for their administrative and technical assistance, and Drs. Claudia Kawas and Maria Corrada-Bravo for their valuable comments.
References
-
Querfurth HW, LaFerla FM (2010) Alzheimer's disease. N Engl J Med 362: 329-344.
-
Mecocci P, Tinarelli C, Schulz RJ, Polidori MC (2014) Nutraceuticals in cognitive impairment and Alzheimer's disease. Front Pharmacol 5: 147.
-
Evans DA, Morris MC, Rajan KB (2014) Vitamin E, memantine, and Alzheimer disease. JAMA 311: 29-30.
-
Maiese K, Chong ZZ, Hou J, Shang YC (2009) The vitamin nicotinamide: translating nutrition into clinical care. Molecules 14: 3446-3485.
-
Sauve AA (2008) NAD+ and vitamin B3: from metabolism to therapies. J Pharmacol Exp Ther 324: 883-893.
-
Herskovits AZ, Guarente L (2013) Sirtuin deacetylases in neurodegenerative diseases of aging. Cell Res 23: 746-758.
-
Hall JA, Dominy JE, Lee Y, Puigserver P (2013) The sirtuin family's role in aging and age-associated pathologies. J Clin Invest 123: 973-979.
-
Niren NM (2006) Pharmacologic doses of nicotinamide in the treatment of inflammatory skin conditions: a review. Cutis 77: 11-16.
-
Gale EA, Bingley PJ, Emmett CL, Collier T (2004) European Nicotinamide Diabetes Intervention Trial (ENDIT): a randomised controlled trial of intervention before the onset of type 1 diabetes. Lancet 363: 925-931.
-
Knip M, Douek IF, Moore WP, Gillmor HA, McLean AE, et al. (2000) European Nicotinamide Diabetes Intervention Trial Group Safety of high-dose nicotinamide: a review. Diabetologia 43: 1337-1345.
-
McCreanor GM, Bender DA (1986) The metabolism of high intakes of tryptophan, nicotinamide, and nicotinic acid in the rat. Br J Nutr 56: 577-586.
-
Marcus R, Coulston AM (1995) Water-soluble vitamins. The vitamin B complex and ascorbic acid. In: Hardman JG, Limbird LE, Molinoff PB, et al. (9th edn), The Pharmacological Basis of Therapeutics, New York, USA, 1555-1571.
-
Petley A, Macklin B, Renwick AG, Wilkin TJ (1995) The pharmacokinetics of nicotinamide in humans and rodents. Diabetes 44: 152-155.
-
Green KN, Steffan JS, Martinez-Coria H, Sun X, Schreiber SS, et al. (2008) Nicotinamide restores cognition in Alzheimer's disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. J Neurosci 28: 11500-11510.
-
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, et al. (2011) The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 7: 263-269.
-
Harrell, Frank (2001) Regression Modeling Strategies with Applications to Linear Models, Logistic Regression and Survival Analysis, Springer, New York, USA.
-
Holm S (1979) A simple sequentially rejective multiple test procedure. Scand J Statistics 6: 65-70.
-
Brady R, Weinman J (2013) Adherence to cholinesterase inhibitors in Alzheimer's disease: a review. Dement Geriatr Cogn Disord 35: 351-363.
-
Gong B, Pan Y, Vempati P, Zhao W, Knable L, et al. (2013) Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-γ coactivator 1α regulated β-secretase degradation and mitochondrial gene expression in Alzheimer's mouse models. Neurobiol Aging 34: 1581-1588.
-
Liu D, Pitta M, Jiang H, Lee JH, Zhang G, et al. (2013) Nicotinamide forestalls pathology and cognitive decline in Alzheimer mice: evidence for improved neuronal bioenergetics and autophagy procession. Neurobiol Aging 34: 1564-1580.
-
Turunc Bayrakdar E, Uyanikgil Y, Kanit L, Koylu E, Yalcin A (2014) Nicotinamide treatment reduces the levels of oxidative stress, apoptosis, and PARP-1 activity in Aβ(1-42)-induced rat model of Alzheimer's disease. Free Radic Res 48: 146-158.
-
Hathorn T, Snyder-Keller A, Messer A (2011) Nicotinamide improves motor deficits and upregulates PGC-1α and BDNF gene expression in a mouse model of Huntington's disease. Neurobiol Dis 41: 43-50.
-
Jia H, Li X, Gao H, Feng Z, Li X, et al. (2008) High doses of nicotinamide prevent oxidative mitochondrial dysfunction in a cellular model and improve motor deficit in a Drosophila model of Parkinson's disease. J Neurosci Res 86: 2083-2090.
-
Holland MA, Tan AA, Smith DC, Hoane MR (2008) Nicotinamide treatment provides acute neuroprotection and GFAP regulation following fluid percussion injury. J Neurotrauma 25: 140-152.
