

Clinical Medical
Reviews and Case Reports
Sequential, Multimodal Treatment of a Refractory Adult-Onset Still's Disease Complicated by Reactive Hemophagocytic Lymphohistiocytosis
Sarah Rosset-Zufferey1*, Karim Gariani1 and Carlo Chizzolini2
1Service of Internal Medicine, Geneva University Hospital, Geneva, Switzerland
2Service of Immunology and Allergy, Geneva University Hospital, Geneva, Switzerland
*Corresponding author: Sarah Rosset-Zufferey, Service de médecine interne générale, Hôpitaux Universitaires de Genève, 4 Rue Gabrielle-Perret-Gentil, 1211 Genève, Switzerland, Tel: +41-79-553-45-61, Fax: +41-22-372-92-35, E-mail: sarah.zufferey@hcuge.ch
Clin Med Rev Case Rep, CMRCR-3-117, (Volume 3, Issue 7), Case Report; ISSN: 2378-3656
Received: April 26, 2016 | Accepted: July 05, 2016 | Published: July 08, 2016
Citation: Rosset-Zufferey S, Gariani K, Chizzolini C (2016) Sequential, Multimodal Treatment of a Refractory Adult-Onset Still's Disease Complicated by Reactive Hemophagocytic Lymphohistiocytosis. Clin Med Rev Case Rep 3:117. 10.23937/2378-3656/1410117
Copyright: © 2016 Rosset-Zufferey S, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
We report the case of a patient with severe adult-onset Still's disease not responding to treatment with high-dose corticosteroids and intravenous immunoglobulins. Partial remission was obtained under therapy associating anakinra and leflunomide but subsequently she developed acute hepatitis and disseminated intravascular coagulation consistent with a reactive hemophagocytic lymphohistiocytosis. The patient was successfully treated with a combination of tocilizumab, cyclosporine and corticosteroids leading to a prolonged remission. This report illustrates the difficulty to treat adult-onset Still's disease complicated by hemophagocytic lymphohistiocytosis and the usefulness of liver biopsy to investigate acute hepatitis in this clinical setting.
Keywords
Adult onset Still's disease, Macrophage activation syndrome, Reactive hemophagocytic lymphohistiocytosis, Hyperferritinemic syndrome, Acute liver injury, Cyclosporine A, Tocilizumab, Anakinra, High dose immunoglobulins
Introduction
Adult onset Still's disease (AOSD) is a rare inflammatory disorder that mainly affects young adults with a slight preponderance for women [1]. The precise etiology and pathogenesis of this disorder remain poorly defined, but it is suggested that the inappropriate production of IL-1 and other pro-inflammatory cytokines, including TNF, IL-6 and IL-18 may play a role in genetically predisposed hosts. For this reason, AOSD may be included in the spectrum of auto-inflammatory disorders [2]. Clinically, most of the patients with AOSD typically present with a spiking fever, classically twice a day, accompanied by a salmon-pink bumpy or flat rash, odynophagia and dysphagia, polyarthritis and myalgia. Other features may include polyserositis, lymphadenopathy, and hepatosplenomegaly [1,2]. Life threatening conditions such as disseminated intravascular coagulation (DIC), thrombotic thrombocytopenic purpura, diffuse alveolar hemorrhage or reactive hemophagocytic syndrome (HS) occasionally occur during the course of AOSD [3].
Hemophagocytic lymphohistiocytosis (HLH), also named macrophage activation syndrome (MAS), can complicate AOSD and may be fatal. The distinction between severe AOSD and HLH may be difficult, since they share common features, especially hepatic injury. The rapid choice of an adequate treatment is essential, but may be complicated by drug hepatotoxicity. This case report illustrates the complexity of refractory AOSD, requiring sequential therapeutic adaptations.
Case
A previously healthy 28-year-old female from Bolivia presented in our department with a two week-history of persistent high fever associated with asthenia, a sore throat and polyarthralgia. During a previous consultation, these symptoms were confirmed and oral penicillin had been prescribed for the suspicion of pharyngitis with no improvement of her symptoms. The rest of the clinical history was irrelevant; particularly the patient had no ocular or urogenital symptoms. She had no significant personal or familial medical history and was not taking any medication. She was a non-smoker and with no history of a high-risk sex encounter or travelling. On physical examination, the patient was febrile at 38.8°C and hemodynamically stable. Bilateral, cervical lymphadenopathy and a generalized macular pruritic fixed rash especially on the trunk and the lower limb were present. The rest of the clinical status was normal. Abnormal laboratory findings included: total white blood cell count of 23 G/l (normal range: 4-11 G/l) with 75% of segmented neutrophils and 15% of band forms, C-reactive protein 398 mg/l (normal range: 0-10 mg/l), ferritin > 7500 mcg/l (normal range: 7-171 mcg/l) with a level of glycosylated ferritin of < 7% (normal range: 50-80%), sedimentation rate 85 mm/h (normal range 0-20 mm/h) and slightly elevated liver transaminases AST 106 U/l and ALT 80 U/l (normal range: < 42 U/l). An infectious origin was suspected in light of the clinical presentation. Urine, blood as well as cerebrospinal fluid cultures were sterile. Serology of HAV, Borrelia burgdorferi, Toxoplasma gondii, Trypanosoma cruzi, Syphilis and Measles, serology and PCR for HBV, HCV, HIV, CMV, EBV, HSV 1, HSV 2 and PCR for HHV 6 showed no active disease. PCR in the nasopharyngeal secretions for common respiratory viruses were negative. Chlamydia and Gonococcus in the urine were negative. The immunologic findings included ANA at 1:160, rheumatoid factor IgM of 89 U, negative ANCA and cryoglobulin and a normal level of immunoglobulin and complement C3-C4. Dermatomyositis was not retained in the absence of auto antibodies, normal CK levels and no signs of myositis on the MRI of the lower limbs. A CT scan of the chest and abdomen and an ultrasound of the abdomen showed no malignancy and no infection. There was a hepatomegaly measuring 20 centimeters. Finally, skin biopsy findings were unspecific, in particular with no arguments for Sweet disease.
The diagnosis of AOSD was made and treatment with oral prednisone (1 mg/kg/day) was initiated. In the absence of clinical and laboratory improvement, high dose intravenous immunoglobulins (IVIG, 2 g/kg of body weight) were introduced 5 days later with no clinical improvement. A combined therapy of anakinra and leflunomide was added and led to a significant clinical improvement and marked reduction in the inflammatory parameters except for a persistently elevated ferritin level (> 7500 mcg/l). However, mild eosinophilia (0.84 G/l) appeared.
This combined therapy was discontinued due to a progressive increase in liver transaminases (AST 252 U/l and ALT 275 U/l), which was suspected to be due to drug toxicity. Tocilizumab (8 mg/kg) was introduced. Transaminases decreased and further clinical improvement was observed but eosinophilia (2.09 G/l) persisted. The TSH was within the normal range. Investigations were performed to identify the cause of eosinophilia including serologies for helminths (a serological panel for Trichinellosis, Toxocariasis, Echinococcosis, Fasciolosis, Schistosomiasis, Filariasis and Anguillulosis) and a Leishmania serology, which were all negative. Only non-pathogenic parasites were found in the stool (Blastocystis hominis, Endolimax nana and Enteromonas hominis).
Seven days after treatment with tocilizumab, high fever, a macular rash and profound asthenia occurred. Laboratory findings showed persistently elevated ferritin and liver enzymes levels (AST 513 U/l, ALT 281 U/l), while thrombocytopenia, prolonged thromboplastin time, a decrease in fibrinogen and marked increased in D-dimers developed indicating disseminated intravascular coagulation (DIC). A transjugular liver biopsy was performed and the histological examination showed portal and lobular lymphocytic infiltration associated with hepatocyte necrosis compatible with reactive hepatitis. There was no evidence for a lymphoproliferative process, hemophagocytosis or a toxic etiology.
The clinical picture, including unremitting fever, liver involvement and DIC lead us to suspect the presence of HLH despite the lack of histological confirmation. A second administration of IVIG (2 g/kg) was with no effect. Cyclosporine A (CsA, 3 mg/kg, bid) was initiated with fortnightly tocilizumab (8 mg/kg) and high dose steroids. Under this therapy the patient showed a considerable improvement and was discharged (see Table 1 for more details about the laboratory values during the hospitalization).
![]()
Table 1: The main laboratory values during the hospitalization.
View Table 1
At 16 months follow-up no recurrence was noted. CsA was stopped after 5 months, prednisone tapered to 5 mg/d, and monthly tocilizumab continued.
Discussion
This case report illustrates the challenge in the diagnosis and treatment of AOSD. Despite prompt and adequate treatment, the patient developed severe complications that lead to question the initial diagnosis and modify treatment.
AOSD remains a diagnosis of exclusion. The differential diagnosis includes infections, neoplastic processes such as lymphomas, systemic autoimmune disorders and primary or reactive HLH [1,3]. In this case, we initially conducted extensive testing to exclude an infectious cause. Serologies and PCR were particularly important to exclude different viruses and Chagas disease, which was suspected because of the origin of the patient. The development of eosinophilia led to the investigation of a parasitic cause. After infectious causes were excluded, a possible neoplasia was investigated, and in particular a hematologic disease such as lymphoma, the most likely cause given the patient's demographics. A complete radiological workup was performed and excluded adenopathy and tumor. Blood smears done under prednisone showed bone marrow stimulation and the BCR-ABL was negative. Finally, concerning immunologic disorders, one of the potential diagnosis was dermatomyositis due to the atypical skin rash and myalgias. It was excluded by negative CK, negative auto-antibodies and the absence of signs of myositis on the MRI. The skin rash was not suggestive of vasculitis. The clinical presentation was not suggestive of spondyloarthritis or of connective tissue diseases.
The diagnosis of AOSD was made according to the Yamaguchi criteria. The patient had 3 of 4 major criteria: fever, leukocytosis and arthralgia. The skin rash was present but atypical because of its fixed and pruritic nature. She had 4 of the 5 minor criteria: a sore throat, cervical lymph node enlargement, liver dysfunction and negative ANA and FR (ANA level of 1:160 and FR of 80 U were considered negative). Only splenomegaly was absent. These criteria have a sensitivity of 96% and a specificity of 92% [4].
High level of serum ferritin was another argument in favor of this diagnosis. The presence of very high ferritin level defines « the hyperferritinemic syndrome » classically reported in four conditions: AOSD, HLH, septic shock, and catastrophic antiphospholipid syndrome (cPAP) [5]. We also found a glycosylated ferritin level (GF) under < 20%. This value appears in the Fautrel and al. criteria for AOSD [6]. In cases of AOSD, the combination of GF levels < 20% with a total ferritin serum 5 times above the upper normal limit has a specificity of 92.9% [7]. The percentage of glycosylated ferritin appears low both in the active phase and during remission, in contrast with the serum ferritin, which changes with the activity of the disease [8]. Interestingly, in our case the serum ferritin levels continued to be high despite treatment with corticosteroids, anakinra and leflunomide and despite a decrease in CRP and clinical improvement (Figure 1). Only the combination of corticosteroids, tocilizumab and CsA resulted in a decrease of the ferritin and clinical improvement.
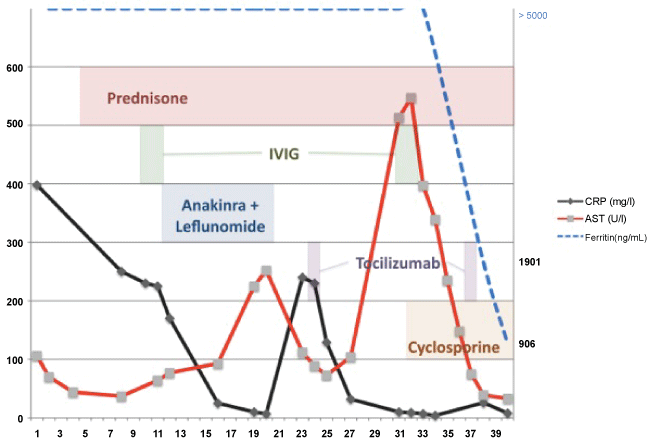
The development of DIC and acute hepatitis was the second biological sign of an uncontrolled disease. The diagnosis of AOSD was reevaluated. A toxic etiology or a reactive HLH were suspected. HLH and AOSD share common features, which make the distinction between these entities challenging. As a consequence of this notable overlap, recognition of HLH complicating AOSD remains difficult, and the combined incidence of these two entities is probably underestimated. A score named the HScore (http://saintantoine.aphp.fr/score/) is currently available and helps to predict the probability of reactive HLH syndrome [9]. Interestingly, the post hoc application of the HS score in our case assessed early during hospitalization was 169, giving a probability of HLH of 52% (see Table 1, labs values at the beginning of the hospitalization), and increased to 223 (see Table 1, labs values at the time of the clinical deterioration), giving a probability of 96.9 % of HLH with the development of biological DIC. It is interesting that, in this situation, the presence of a haemophagocytosis in the bone marrow biopsy would have increased the probability by less than 3% (99.6%). Thus, the HS score appears to be a useful adjunct in everyday practice helping in distinguishing severe AOSD from HLH, thus perhaps avoiding unnecessary diagnostic procedures.
To investigate hepatic injury, a hepatic biopsy was performed. It excluded hepatotoxicity due to the use of anakinra [10], leflunomide or tocilizumab [11]. The biopsy showed reactive hepatitis, which we interpreted as a sign of an uncontrolled disease. The literature remains scarce about the hepatic biopsy findings in cases of reactive HLH, but the absence of hemophagocytosis does not exclude this diagnosis [12,13].
No response to corticosteroids defines refractory AOSD, requiring the introduction of DMARDs. Methotrexate is the most commonly used cDMARDs, but leflunomide was given because it is less hepatotoxic. The off-label administration of biological DMARDs such as anti-TNFs (infliximab, adalinumab, etanercept) and anti-IL1 (anakinra) is described in several case reports [1]. Anti-IL6 (tocilizumab) is also a potential option, maybe explained by the high levels of IL-6 found in patients with AOSD [14]. In conclusion, switching from one biological agent to another, each targeting a different cytokine may suggest that the effective target varies amongst patients. Predicting which treatment will be the most effective for each patient is a challenge.
Established recommendations for the management of HLH complicating AOSD are lacking. It has been reported that cyclosporine associated with corticosteroids has been successful in MAS secondary to juvenile rheumatoid arthritis [5]. The efficacy of IVIG is controversial and etoposide can be considered in cases refractory to treatment with cyclosporine [15].
In conclusion, it remains difficult to predict the course of AOSD. Some patients experience a single mild episode, while others may present life-treating conditions. The present case illustrates these difficulties and the need to promptly adapt treatment according to the changes in the severity of clinical symptoms and biological parameters. After the failed response to corticosteroids, IVIG, anakinra and lefunomide, the co-administration of tocilizumab, CsA, and corticosteroids lead to prolonged remission.
Conflict of Interests
The authors declare no conflict of interests.
References
-
Efthimiou P, Kontzias A, Ward CM, Ogden NS (2007) Adult-onset Still's disease: can recent advances in our understanding of its pathogenesis lead to targeted therapy? Nat Clin Pract Rheumatol 3: 328-335.
-
Efthimiou P, Georgy S (2006) Pathogenesis and management of adult-onset Still's disease. Semin Arthritis Rheum 36: 144-152.
-
Efthimiou P, Kadavath S, Mehta B (2014) Life-threatening complications of adult-onset Still's disease. Clin Rheumatol 33: 305-314.
-
Yamaguchi M, Ohta A, Tsunematsu T, Kasukawa R, Mizushima Y, et al. (1992) Preliminary criteria for classification of adult Still's disease. J Rheumatol 19: 424-430.
-
Rosário C, Zandman-Goddard G, Meyron-Holtz EG, D'Cruz DP, Shoenfeld Y (2013) The hyper ferritinemic syndrome: macrophage activation syndrome, Still's disease, septic shock and catastrophic antiphospholipid syndrome. BMC Med 11: 1-11.
-
Fautrel B, Zing E, Golmard JL, Le Moel G, Bissery A, et al. (2002) Proposal for a new set of classification criteria for adult-onset still disease. Medicine (Baltimore) 81: 194-200.
-
Fautrel B, Le Moël G, Saint-Marcoux B, Taupin P, Vignes S, et al. (2001) Diagnostic value of ferritin and glycosylated ferritin in adult onset Still's disease. J Rheumatol 28: 322-329.
-
Vignes S, Le Moël G, Fautrel B, Wechsler B, Godeau P, et al. (2000) Percentage of glycosylated serum ferritin remains low throughout the course of adult onset Still's disease. Ann Rheum Dis 59: 347-350.
-
Fardet L, Galicier L, Lambotte O, Marzac C, Aumont C, et al. (2014) Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol 66: 2613-2620.
-
Taylor SA, Vittorio JM, Martinez M, Fester KA, Lagana SM, et al. (2016) Anakinra-Induced Acute Liver Failure in an Adolescent Patient with Still's Disease. Pharmacotherapy 36: e1-4.
-
Drepper M, Rubbia-Brandt L, Spahr L (2013) Tocilizumab-Induced Acute Liver Injury in Adult Onset Still's Disease. Case Reports Hepatol 2013: 964828.
-
Lim KB, Schiano TD (2011) Still disease and the liver-an underappreciated association. Gastroenterol Hepatol (N Y) 7: 844-846.
-
Larroche C, Ziol M, Zidi S, Dhote R, Roulot D (2007) Liver involvement in hemophagocytic syndrome. Gastroenterol Clin Biol 31: 959-966.
-
de Boysson H, Février J, Nicolle A, Auzary C, Geffray L (2013) Tocilizumab in the treatment of the adult-onset Still's disease: current clinical evidence. Clin Rheumatol 32: 141-147.
-
Emmenegger U, Frey U, Reimers A, Fux C, Semela D, et al. (2001) Hyperferritinemia as indicator for intravenous immunoglobulin treatment in reactive macrophage activation syndromes. Am J Hematol 68: 4-10.
