

Clinical Medical
Reviews and Case Reports
The Use of Macitentan in the Treatment of Out-of-Proportion Pulmonary Hypertension in Chronic Obstructive Pulmonary Disease: Case Report
Luca Fallavollita1*, Paolo Spinaci2 and Pietro Scendoni1
1Italian National Research Center on Aging, Fermo, Italy
2Unit of Pneumology, Osimo Hospital, Italy
*Corresponding author: Dr. Luca Fallavollita, Unit of Cardiology, Italian National Institute of Research on Aging INRCA, 6 Canale street, Fermo, Italy, Tel: +39-347-9331369, E-mail: loufall79@katamail.com
Clin Med Rev Case Rep, CMRCR-3-147, (Volume 3, Issue 12), Case Report; ISSN: 2378-3656
Received: October 26, 2016 | Accepted: December 12, 2016 | Published: December 14, 2016
Citation: Fallavollita L, Spinaci P, Scendoni P (2016) The Use of Macitentan in the Treatment of Out-of-Proportion Pulmonary Hypertension in Chronic Obstructive Pulmonary Disease: Case Report. Clin Med Rev Case Rep 3:147. 10.23937/2378-3656/1410147
Copyright: © 2016 Fallavollita L, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Keywords
Macitentan, Out of proportion pulmonary hypertension, Echocardiography, Right heart catheterization
Introduction
Pulmonary hypertension (PH) is an important complication of chronic pulmonary disease, especially of chronic obstructive lung disease (COPD) [1]. The exact prevalence of PH in COPD is unknown. PH is defined as an increase in mean pulmonary arterial pressure (PAPm) > 25 mmHg at rest as assessed by right heart catheterization [2]. In most cases, PH in COPD patients is usually mild to moderate [1]. Only a small proportion of this cohort may present with severe pulmonary hypertension [3-5]. This condition with poor prognosis is characterized by exertional dyspnea disproportionate to pulmonary function tests, low carbon monoxide diffusion capacity and rapid decline of arterial oxygenation upon exercise. The current clinical classification of pulmonary hypertension includes PH secondary to COPD in subgroup 3, called PH associated to lung diseases and/or hypoxia [6].
The pathophysiology of PH associated with COPD is not well established [1]. Echocardiography is recommended for the non-invasive diagnostic assessment of suspected PH in patients with lung disease [7]. Lung function test, chest high resolution CT scan, caridiopulmonary exercise testing and ventilation-perfusions play a central role in recognition of PH in COPD [7].
Right heart catheterization is not routinely recommended in the assessment of patients with COPD and its indication is reserved for selected cases such as severe PH suspected by noninvasive measure and/or clinical worsening and progressive exercise limitation disproportionate to ventilator impairment [8].
In patients with COPD and associated PH, the respiratory disease should be optimally treated according to existing guidelines [9]. Treatment with drugs used to treat PAH (prostanoids, endothelin receptor antagonists, phosphodiesterase-5-inhibitors and guanylate cyclase stimulators) is generally discouraged due to lack of consistent efficacy observed in the majority of clinical trials conducted so far [10]. However, clinical benefit with PAH therapy has been reported in isolated cases [4,3,11] and the use of target PH therapy might be useful in COPD patients with severe PH, where the predominant problem is evidently vascular disease and management of their clinical illness is challenging [12].
Macitentan is the newest dual endothelin receptor antagonist approved to long-term treatment of pulmonary arterial hypertension (PAH). However, there is no evidence for its routine use in severe pulmonary hypertension (PH) in COPD patients. We report the first case of severe PH in a patient with chronic obstructive pulmonary disease who favorably responded to macitentan.
Case Report
A 70-year-old woman with a 20 pack-year smoking history, coronary artery disease, hypertension and chronic obstructive pulmonary disease (COPD) presented at our cardiology clinic with a 3-month history of worsening dyspnea on exertion and bilateral lower extremity edema.
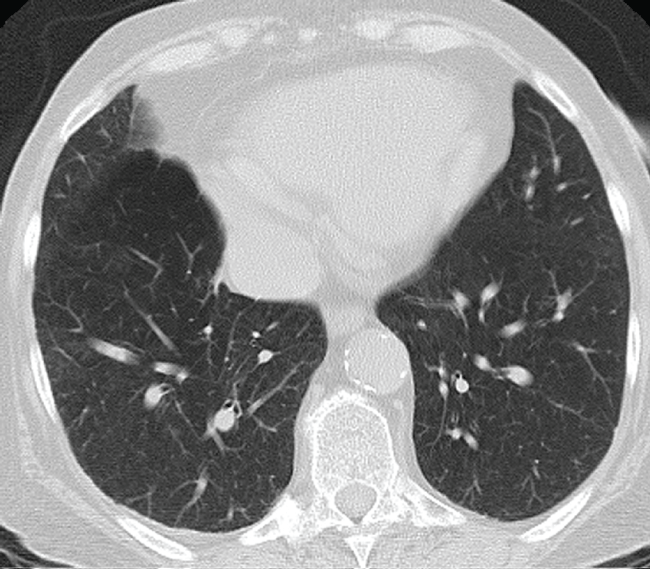
On admission she had severe dyspnea and she has been classified in WHO class IV. Physical examination revealed increased jugular venous pressure, blood pressure 110/60 mmHg and heart rate of 100 bpm. Pulmonary auscultation revealed decreased vesicular breath sounds and bilateral basal sizzling rales. The abdomen had no ascites, and there was discrete edema in the lower limbs. Laboratory tests have shown: marked increase of BNP (3785 pg/mL) and slight increase of troponin I (0.05 ng/mL), D-dimer (313 ng/mL), Creatinine 1.47 mg/dL. Arterial Blood Gas Analysis parameters were: pH 7.36, pO2 42.7 mmHg, pCO2 43.4 mmHg, HCO3 24.3 mEql/l, SaO2 77%. Screening tests for hepatitis were negative. Abdominal ultrasound showed no portal hypertension. The electrocardiogram showed sinus rhythm a heart rate of 97 bpm, incomplete right bundle branch block, premature ventricular contraction, QTc 474 msec. Chest radiography revealed lung markings. Transthoracic echocardiography (TTE) showed left ventricle within normal limits, normal ejection fraction (LVEF 50%) and mild diastolic dysfunction (E/A 0.77; E/E’ 10); dilation right ventricle and right atrium, paradoxical interventricular septal motion, decrease of tricuspid anular plane systolic excursion (TAPSE 12 mm), severe maximal tricuspidal regurgitation velocity (TR Vmax) 4.48 m/s (Figure 1), the inferior vena cava diameter slightly increased with a small tidal variability. Pulmonary artery pressure was estimated to be at 95 mmHg. Thoracic Angio-Computed Tomography showed no evidence of pulmonary embolism. Rheumatologic laboratory testing was negative. High resolution computer tomography (HRCT) exhibited severe emphysematous change (Figure 2).
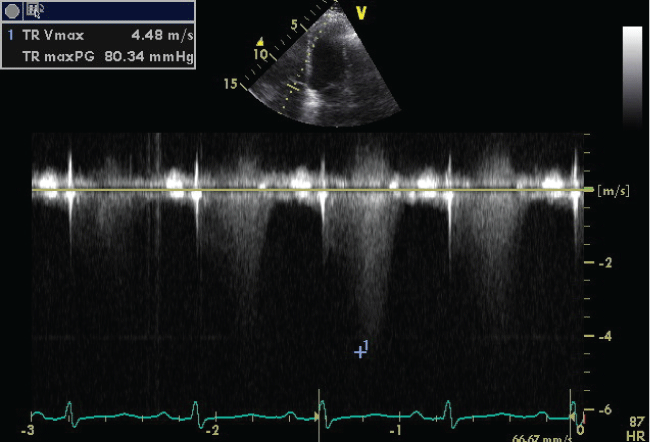
.
Figure 2: High-resolution tomography scan shows a pattern of pulmonary emphysematous bubbles.
View Figure 2
The six minutes walking test (6MWT) was not performed because of impaired clinical conditions. Pulmonary function test (PFT) showed moderate decrease in forced expiratory volume in 1 second (FEV1 = 1,33 L; 66% of predicted), in forced vital capacity (FVC = 2,35 L; 98% of predicted), in FEV1/FVC (57% of predicted) and in diffusing capacity of the lung for carbon monoxide (DLCO 5.4 mL/mmHg/min; % DLCO 26% of predicted; DLCO/VA 1.59 mL/mmHg/min/L). The patient received oxygen supplementation via nasal catheter adjusted according to the blood gases, diuretics (furosemide 60 mg/day; canrenone 50 mg/day), acetylsalicylic acid (100 mg/day), bisoprolol (1.25 mg/day) and bronchodilatators (fluticasone 92 mcg/day and vilanterol 22 mcg/day) and previous therapy (atorvastatin 40 mg/day; ticagrelor 90 mg bid day). As there was no improvement with standard treatment, right heart catheterization was performed showing: right atrial pressure (RAP) 10 mmHg, right ventricular pressure (RVP) 15 mmHg, mean pulmonary artery pressure (mPAP) 54 mmHg, pulmonary capillary wedge pressure (PCWP) 14 mmHg, cardiac index (CI) 1.91/min/mq, pulmonary vascular resistance (PVR) 721 dyn s/cm2. On the basis of RHC, we diagnosed out-of-proportion pulmonary hypertension. Hence, given the gravity of clinical condition and the refractory to therapy, we started an off-label treatment with macitentan 10 mg per day. One month later, the patient tolerated treatment well without acute side effects and we observed clinical, echocardiography and laboratory improvement. Particularly, the patient had no exertional dyspnea, she had less peripheral edema and the functional class improved from class IV to class II. TTE showed decreased of TR Vmax (3.65 m/s) (Figure 3) with improvement of pulmonary artery pressure (approximately 65 mmHg) and tricuspid anular plane systolic excursion (TAPSE 17 mm). The BNP decreased significantly (153 pg/mL). The patient was finally able to perform 6MWT. She reached the distance of 248 m without significant desaturation (SaO2 95%) achieving 11/20 points in Borg scale at the end of the test. Liver profile laboratory and hemoglobin were normal during follow-up.
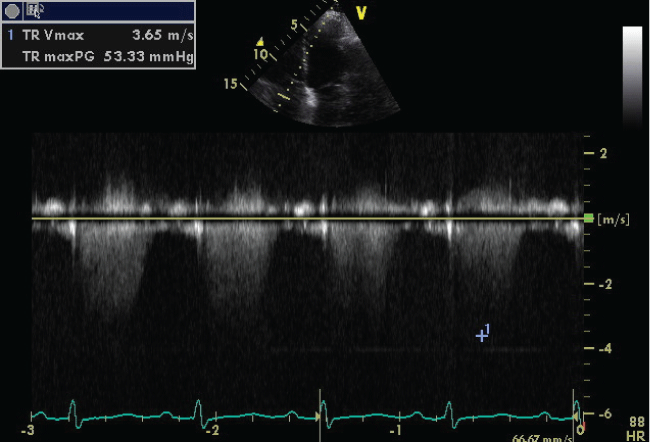
.
Figure 3: Transthoracic echocardiography scan shows decrease value of TR Vmax after macintent therapy.
View Figure 3
Discussion
Pulmonary hypertension (PH) is a common complication in the population of patients with chronic obstructive pulmonary disease (COPD) [1]. In these patients, PH is usually mild to moderate [1]. Only in a small percentage of patients with COPD (< 5%) develops extremely high pulmonary artery pressure [3-5]. Clinically this condition PH is characterized by severe pulmonary hypertension, hypoxemia, low diffusion capacity of the lung for carbon monoxide and moderate airway obstruction [12]. Moreover, the clinical course tends to be worse, acute exacerbations of COPD are said to be more common and more severe and the right heart failure tend to dominate the clinical picture [13]. The prognosis of these patients is very poor with a 5-year survival rate of 10%, compared with rates of 60% in patients with less severe PH and almost 90% in patients without PH [12,14,15]. The mechanism that produces severe PH in a small proportion of COPD patients is likely complex and still unclear [1]. Recent studies provide evidence on the important role of the endothelial cell and its derived mediators in the pathogenesis of PH associated with respiratory disease [16,10]. Particularly, endothelial dysfunction plays a key role in the pathogenesis of PH in COPD [16,10] and is associated with impaired release of endothelium-derived vasodilating (nitric oxide, prostacyclin) and vasoconstrictive agents (endothelin1) [12]. Endothelin-1(ET-1) is the most potent vasoconstrictor and it is a key mediator of vasculopathy. ET-1 promotes vasoconstriction, smooth muscle cell proliferation and adventitia fibrosis, inducing a vascular narrowing. The use pulmonary vasodilators (prostanoids, endothelin-1 receptor antagonist and phosfpdiesterase-5 inhibitors) could be considered in this setting of patients. In an Italian study recently published sildenafil reduced pulmonary vascular resistance and improved the body mass index, airflow obstruction, dyspnea, exercise capacity index and quality of life, without a significant effect on gas exchange [17]. Previously, in a series of cases, PAH specific drugs have improved hemodynamic parameters in COPD patients with severe PH [7]. However, there are not randomized, double blind studies performed in patients with COPD and PH and the use of PAH specific drugs remains controversial. In summary, the use of PAH specific drugs is not recommended in these patients [6], although their use may be justified where the predominant problem is evidently vascular disease and management of their clinical illness is challenging [12].
In this report, we describe the case of a patient with COPD and severe pulmonary hypertension, who dramatically improved after treatment with macitentan. Macitentan is the most recently approved dual endothelin-receptor antagonist (ERA) for the treatment of pulmonary artery hypertension. Compared to other ERAs, it demonstrates superior receptor-binding properties, with improved tissue penetration and a longer duration of action allowing for once-daily dosing [18]. Also macitentan displays higher efficacy, lesser adverse effects and drug interactions [19]. In the recently published SERAPHIN study [12], treatment with macitentan significantly reduced morbidity and mortality among patients with pulmonary arterial hypertension (in group 1). Moreover, in this setting of patients macitentan has been shown to improve functional class, exercise tolerance and hemodynamic parameters.
In our case, macitentan improved the general condition of the patient, exercise capacity, echocardiographic parameters and reduced symptoms. Also macitentan was safe in our case. We hypothesize that the clinical benefits induced by macitentan in this patient can be attributes to its vasodilator and anti-proliferative actions at the pulmonary vascular level. In this regard, the case report suggests a potential role of macitentan in the treatment of out-of-proportion pulmonary hypertension patients. However, further and larger studies are needed to better define the safety and efficacy of macitentan in this clinical setting. Limitations of this case report include: the short period of observation (at the time of the drafting of the manuscript, the patient must still be re-evaluated at the half-year follow-up); the clinical response was not confirmed from hemodynamic point of view (the patient refused to repeat RHC following PH specific therapy); finally, we have not performed the HIV test, to exclude a form of PH associated with HIV (we judged the patient a very low risk for this clinical condition).
Conclusion
PH is a common complication in patients with COPD. In most cases, PH is generally mild to moderate. Only a small proportion of patients develop severe PH, known as out-of-proportion. Out-of-proportion is a grey area of medicine and the use pulmonary vasodilators remains controversial. In our case, macitentan significantly improved general condition of the patient, reduced symptoms and improved echocardiographic findings. Further investigation is needed to better elucidate the potential therapeutic role of pulmonary vasodilators in this clinical setting.
Conflict of Interest
The authors have no conflict of interest to disclose.
References
-
Chaouat A, Naeije R, Weitzenblum E (2008) Pulmonary hypertension in COPD. Eur Respir J 32: 1371-1385.
-
Hoeper MM, Bogaard HJ, Condiffe R, Frantz R, Khanna D, et al. (2013) Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol 62: D42-D50.
-
Lasota B, Skoczynski S, Mizia-Stec K, Pierzchala W (2013) The use of iloprost in the treatment of 'out of proportion' pulmonary hypertension in chronic obstructive pulmonary disease. Int J Clin Pharm 35: 313-315.
-
Hegewald MJ, Elliott CG (2009) Sustained improvement with iloprost in a COPD patient with severe pulmonary hypertension. Chest 135: 536-537.
-
Harari S (2012) Out-of-proportion pulmonary hypertension: a paradigm for rare diseases. Chest 142: 1087-1088.
-
Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, et al. (2015) 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J 46: 903-975.
-
Girard A, Jouneau S, Chabanne C, Khouatra C, Lannes M, et al. (2015) Severe pulmonary hypertension associated with COPD: hemodynamic improvement with specific therapy. Respiration 90: 220-228.
-
Seeger W, Adir Y, Barberŕ JA, Champion H, Coghlan JG, et al. (2013) Pulmonary hypertension in chronic lung disease. J Am Coll Cardiol 62: D109-D116.
-
http:/www.goldcopd.org.
-
Barberr JA, Blanco I (2015) Management of Pulmonary Hypertension in Patients with Chronic Lung Disease. Curr Hypertens Rep 17: 62.
-
Cottin V, Khouatra C, Lazor R, Canu P, Cordier JF (2009) Pulmonary hypertension therapy and COPD: still many questions to be answered. Eur Respir J 33: 450-452.
-
Zakynthinos E, Daniil Z, Papanikolaou J, Makris D (2011) Pulmonary hypertension in COPD: pathophysiology and therapeutic targets. Curr Drug Targets 12: 501-513.
-
Gross NJ (2010) The COPD pipeline VI. COPD 7: 383-385.
-
Thabut G, Dauriat G, Stern JB, Logeart D, Levy A, et al. (2005) Pulmonary hemodynamics in advanced COPD candidates for lung volume reduction surgery or lung transplantation. Chest 127: 1531-1536.
-
Scharf SM, Iqbal M, Keller C, Criner G, Lee S, et al. (2002) Hemodynamic characterization of patients with severe emphysema. Am J Respir Crit Care Med 166: 314-322.
-
Peinado VI, Pizarro S, Barberŕ JA (2008) Pulmonary vascular involvement in COPD. Chest 134: 808-814.
-
Vitulo P, Stanziola A, Confalonieri M, Libertucci D, Oggionni T, et al. (2016) Sildenafil in severe pulmonary hypertension associated with chronic obstructive pulmonary disease: A randomized controlled multicenter clinical trial. J Heart Lung Transplant.
-
Kholdani CA, Fares WH, Trow TK (2014) Macitentan for the treatment of pulmonary arterial hypertension. Vasc Health Risk Manag 10: 665-673.
-
Khadka A, Singh Brashier DB, Tejus A, Sharma AK (2015) Macitentan: An important addition to the treatment of pulmonary arterial hypertension. J Pharmacol Pharmacother 6: 53-57.
