

Clinical Medical Reviews and Case Reports
Acute Liver Failure Secondary to AL Amyloidosis
Brian J Horner1, Ryan Craig2, Estelle S Harris3, Tibor Kovacsovics4 and Juan F Gallegos-Orozco1*
1Division of Gastroenterology, Hepatology, and Nutrition, Department of Internal Medicine, University of Utah, USA
2Department of Pathology, University of Utah, USA
3Division of Pulmonary and Critical Care Medicine, Department of Internal Medicine, University of Utah, USA
4Division of Hematology and Oncology, Department of Internal Medicine, University of Utah, USA
*Corresponding author:
Juan F Gallegos-Orozco MD, Division of Gastroenterology, Hepatology, and Nutrition, Department of Internal Medicine, University of Utah School of Medicine, 30 N 1900 E, Room 4R118, Salt Lake City, UT 84132, USA, E-mail: juan.gallegos@hsc.utah.edu
Clin Med Rev Case Rep, CMRCR-4-157, (Volume 4, Issue 2), Case Report; ISSN: 2378-3656
Received: November 01, 2016 | Accepted: February 25, 2017 | Published: February 28, 2017
Citation: Horner BJ, Craig R, Harris ES, Kovacsovics T, Gallegos-Orozco JF (2017) Acute Liver Failure Secondary to AL Amyloidosis. Clin Med Rev Case Rep 4:157. 10.23937/2378-3656/1410157
Copyright: © 2017 Horner BJ, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
AL amyloidosis, the most common form of amyloidosis, involves the extracellular deposition of immunoglobulin light chain protein fibrils, usually in the setting of a plasma cell dyscrasia. Amyloid deposition can occur in any organ system, including the gastrointestinal tract and liver. In autopsy studies, liver involvement in AL amyloidosis is common but usually has minimal and nonspecific clinical manifestations. Acute liver failure from AL amyloidosis is rare and has only been noted in a few individual case reports. Here, we report a case of AL amyloidosis, likely due to a plasma cell dyscrasia, presenting as rapid development of acute liver failure.
Keywords
Acute liver failure, AL amyloidosis, Monoclonal gammopathy, Plasma cell dyscrasia
Introduction
AL amyloidosis, the most common form of amyloidosis, involves the extracellular deposition of immunoglobulin light chain protein fibrils, usually in the setting of a plasma cell dyscrasia. Amyloid deposition can occur in any organ system, including the gastrointestinal tract and liver. Previously published reports have indicated liver involvement in AL amyloidosis to be fairly common, with a prevalence of 62-90% [1]. Despite this, clinical manifestations of hepatic involvement are minimal and acute liver failure is rare. Here, we report a case of AL amyloidosis, presenting as acute liver failure. Post-mortem examination of bone marrow revealed the presence of a plasma cell dyscrasia, which is the presumed etiology of the AL amyloidosis in this case.
Case Report
A 73-year-old woman was initially admitted to an outside hospital with complaint of edema and increased abdominal girth. Her past medical history was significant only for cholecystitis, for which she underwent cholecystectomy distantly, and hypothyroidism that was well controlled with thyroid hormone replacement.
Laboratory evaluation at that time revealed serum sodium 129 mmol/L, serum total bilirubin 5.7 mg/dL, AST 94 U/L, ALT 31 U/L, albumin 2.0 g/dL, platelet count 254 (× 103/uL) and INR 3.0. Computed tomography of the abdomen showed small volume ascites, a mildly enlarged liver with steatosis, and mild splenomegaly. Abdominal magnetic resonance imaging with magnetic resonance cholangiopancreatography revealed surgically absent gallbladder, no substantial common bile duct dilatation or obstruction, a fatty appearing liver without mass, and moderate volume ascites. Serology was negative for any acute or chronic viral hepatitis infection as were studies for autoimmune hepatitis, primary biliary cholangitis, alpha-one antitrypsin deficiency, and hemochromatosis.
The patient underwent liver biopsy. No significant portal or lobular inflammation or necrosis was seen. The lobules were distorted by diffuse, lightly basophilic, deposits and the hepatocytes were compressed and atrophic, with no remaining residual lobular or central venous architecture. The reticulin stain highlighted portal fibrous tissue and showed the residual reticulin network surrounding the focal cords of hepatocytes. A trichrome stain highlighted the portal tracks and showed light staining of the extensive deposits. No findings of cirrhosis were present. No steatosis, hepatocyte ballooning, nodular regeneration, parenchymal cholestasis was seen. There was no significant iron deposition demonstrated on an iron stain. The liver biopsy was thought consistent with possible amyloidosis based on the presence of a positive Congo red stain, but confirmatory data for such a diagnosis was not available at the time of the initial histological review.
A diagnostic paracentesis was performed indicating the presence of spontaneous bacterial peritonitis (SBP) based on ascites fluid cell count. Treatment was begun with IV ceftriaxone and she was discharged on oral ciprofloxacin. Outpatient follow up with local gastroenterology/hepatology and hematology services was arranged. Three weeks later, prior to receiving any outpatient follow-up, she presented again to the outside hospital with grade III hepatic encephalopathy. Her coagulopathy and elevated serum liver injury tests also persisted, thus meeting American Association for the Study of Liver Diseases (AASLD) diagnostic criteria for acute liver failure. She was subsequently transferred to our institution for further evaluation and management.
Further history was obtained from the patient and her family. She had no personal history of diabetes, hypertension, hyperlipidemia, or metabolic syndrome. She had no prior history of anemia or any other known hematologic abnormalities. She had no personal or family history of malignancy (including no history of hematologic malignancy). She had no recent or past use of herbal or dietary supplements, tobacco, alcoholor illicit/psychedelic substances.
Additional work-up for etiologies of her acute liver failure and an underlying chronic liver disease was undertaken. Serologic studies for hepatitis A, hepatitis B, hepatitis C, hepatitis E, Epstein-Barr virus, cytomegalovirus, and varicella zoster virus were negative. Antinuclear, antismooth muscle, and anti-liver-kidney microsomal antibodies were negative. Antimitochondrial antibody was negative. Ceruloplasmin was normal. Quantitative immunoglobulin measurements were performed revealing IgA 38 mg/d (reference range 68-378 mg/dL), IgM 10 mg/dL (reference range 60-263 mg/dL), IgG 2300 mg/dL (reference range 768-1632 mg/dL).
Additional work-up for amyloidosis was undertaken. Serum protein electrophoresis (SPEP) and measurement of serum kappa and lambda light chains was performed. SPEP revealed an M-spike that was confirmed to be monoclonal IgG kappa by immunoelectrophoresis. Serum kappa free light chains were elevated at 142 mg/dl (reference range 0.33-1.94 mg/dL) and the kappa/lambda free light chain ratio was also elevated at 135.24 (reference range 0.26-1.65). Subtle echocardiographic evidence of longitudinal strain on the intraventricular septum was noted, raising suspicion for cardiac amyloidosis. Liquid chromatography tandem mass spectrometry (LC/MS) performed on peptides extracted from the index liver biopsy specimen had a profile consistent with AL (kappa-type) amyloidosis. The combination of these findings was highly suggestive of AL amyloidosis.
Bone marrow biopsy was performed. The marrow aspirates and core biopsy yielded only minimal material, thus limiting definitive quantitative results on the histologic exam. However, cell count and differential of the aspirate specimen yielded 2.0% plasma cells. Flow cytometric analysis of the bone marrow aspirate demonstrated a CD56 positive monoclonal plasma cell population (approximately 0.31% of the leukocytes), and was indicative of a plasma cell dyscrasia. Chromosomal analysis of bone marrow tissue revealed a normal female chromosome complement. Bone marrow core biopsy contained mostly cortical bone, prohibiting further diagnostic interpretation.
The patient's clinical condition rapidly worsened over 4 days following her admission to our hospital. Her encephalopathy worsened necessitating endotracheal intubation and mechanical ventilation for airway protection. She developed acute oliguric renal failure and required initiation of continuous hemodialysis. She developed disseminated intravascular coagulation as evidenced by a platelet count that declined to 99 × 103/uL, an INR that rose to 5.5, an elevated aPTT of 61 seconds, and a low fibrinogen of 107 mg/dL. This was managed supportively with transfusion of packed red blood cells, fresh frozen plasma, and platelets. The worsening coagulopathy also led to spontaneous hemorrhage at the bone marrow biopsy sites and subsequent circulatory shock that required vasopressor support. The likely systemic amyloidosis precluded consideration of definitive therapy for her liver or kidney failure with solid organ transplant, and her poor clinical and functional status precluded any significant empiric corticosteroid use or chemotherapy for management of the amyloidosis (or possible underlying myeloma). The patient's family subsequently chose to discontinue invasive and advanced therapies in favor of a palliative approach, and she ultimately died of multiorgan system failure.
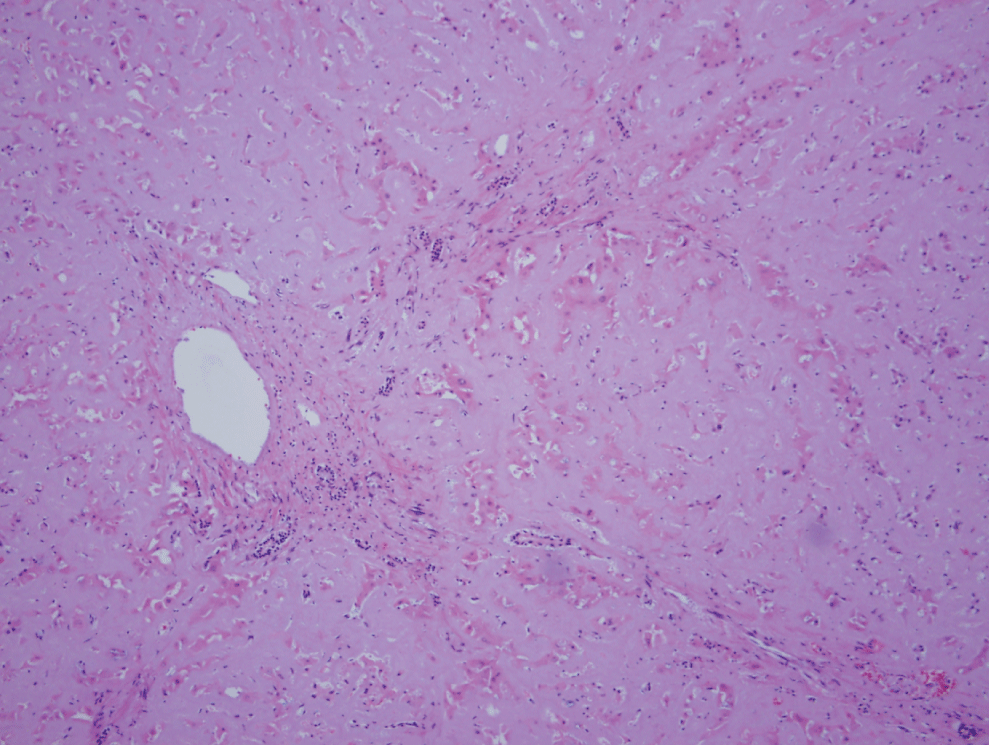
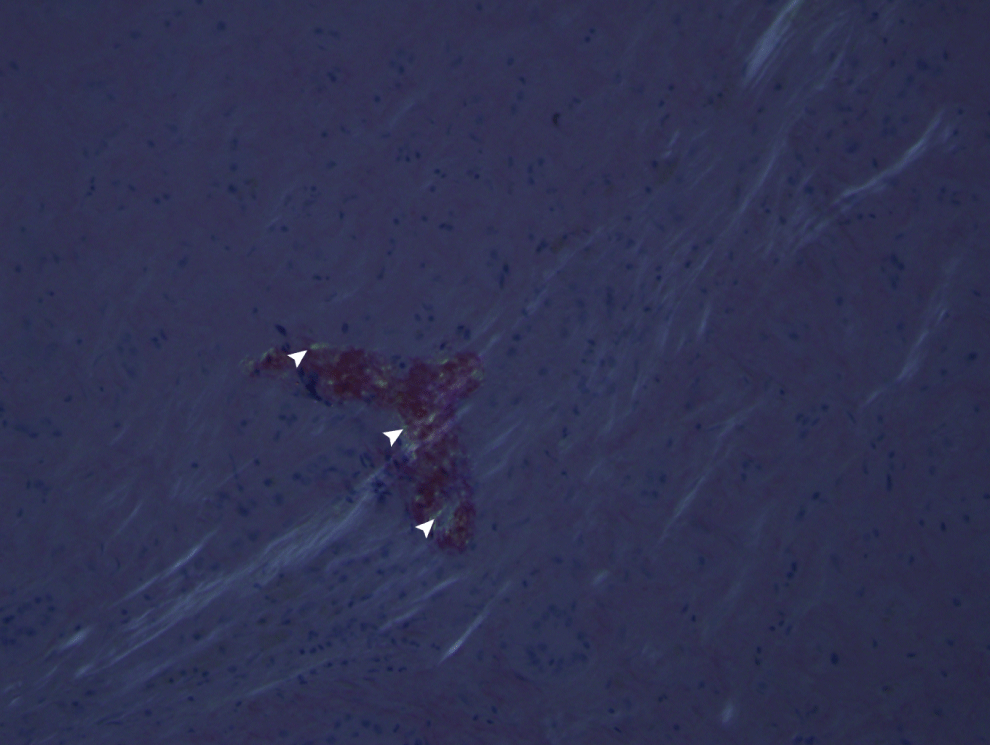
An autopsy was subsequently performed. Grossly, severe jaundice with an extremely large and firm liver, weighing 3500 g was found. Microscopically, global and extensive amyloid deposition was noted throughout the liver in both vessels and the hepatic parenchyma. Normal liver architecture had been severely distorted secondary to the extensive amyloid deposition, and extensive periportal lymphocytic inflammation was also present (Figure 1). Congo red staining was positive for amyloid, exhibited by green birefringence under polarization (Figure 2). Similar to the liver, amyloid deposition was seen in both the vessels and parenchyma of the spleen and kidneys, and there was diffuse deposition of amyloid present within vessels of the heart, adrenal glands, thyroid gland, pancreas, lung, and throughout the GI tract. Bone marrow taken post-mortem showed hypercellularity for age (90%) with trilineage hematopoiesis and an abnormally high percentage of plasma cells, which was felt to be highly suggestive of an underlying plasma cell dyscrasia.
.
Figure 1: H&E stain of liver (magnification x20). Normal liver architecture nearly completely replaced by amyloid deposition as well as presence of extensive periportal lymphocytic inflammation.
View Figure 1
.
Figure 2: Congo red staining of Liver (magnification x20). Green birefringence (arrowheads) under polarization indicative for the presence of amyloid.
View Figure 2
Discussion
The prevalence of liver involvement in AL amyloidosis, as assessed by autopsy studies [1] is relatively common, but manifestation of any symptoms referable to liver involvement is rare. The most common presenting symptoms of hepatic amyloid involvement are fatigue and weight loss, and the most common objective findings of hepatomegaly and elevated alkaline phosphatase are equally nonspecific [1,2]. However, the presence of hepatic amyloidosis portends a poor prognosis. The largest available case series of hepatic amyloidosis, consisting of 98 patients, reports a median survival of only 8.5 months [3].
Despite the high prevalence of hepatic involvement with AL amyloidosis, the development of acute liver failure from this entity is rare. Norero, et al. have previously published a review identifying only 7 published patient cases in the past 30 years of acute liver failure associated with AL amyloidosis [4]. In all of these cases, patients initially presented with relatively nonspecific symptoms such as jaundice, hepatomegaly, anasarca, or weight loss and subsequently developed fulminant liver failure over 2-9 weeks from the time of initial presentation, all thus meeting AASLD acute liver failure diagnostic criteria with encephalopathy, coagulopathy, and hepatic injury in an illness of less than 26 weeks' duration [5]. The patient in our case had a similar presentation to previously published cases with nonspecific signs and symptoms of hyperbilirubinemia, elevated liver injury tests, and anasarca. However, her case is distinguished from others by a much more rapid development of, and clinical decline from, acute liver failure, with just 7 days elapsing from the development of liver failure to her death despite all available supportive measures.
The precipitous decline in this patient's case raises the question of a concomitant hepatic disease process or physiologic insult that might have worsened her clinical status, and particularly her hepatic function, but none could be identified. There were no significant changes or additions to this patient's medication regimen to suggest a superimposed drug induced liver injury. She did not have any stigmata of gastrointestinal bleeding. There was no evidence of portal or hepatic vein thrombosis, and she had not been persistently hypotensive prior to presentation to suggest an ischemic hepatic injury. She was treated for SBP at the time of her initial presentation, was continued on antibiotics when she presented in acute liver failure, and had a repeat paracentesis with cell count and culture that did not have evidence of persistent or untreated peritonitis. There were no objective findings of other systemic infection.
While underlying multiple myeloma was suspected, the patient did meet the current International Myeloma Working Group (IMWG) diagnostic criteria for active multiple myeloma [6]. However, there was ample evidence to support diagnosis of a plasma cell dyscrasia. The bone marrow biopsy showed 2.0% plasma cells by morphology and a monoclonal plasma cell population by flow cytometry. Post-mortem bone marrow examination was also hypercellular with a high percentage of plasma cells. Further, this patient had multiple organs with amyloid infiltration, positive Congo red staining for amyloid in the liver, demonstration of kappa light-chain amyloid by mass spectrometry, and evidence of monoclonal plasma cell proliferation on serum and bone marrow analyses, thus meeting IMWG diagnostic criteria for systemic AL amyloidosis [6].
This patient's final clinical and anatomic diagnoses remain acute liver failure from AL amyloidosis leading to subsequent multi-organ system failure and systemic amyloidosis with global parenchymal involvement of liver and spleen and vascular involvement of heart, kidney, adrenal, thyroid, lung, esophagus, stomach, bowel, pancreas, and uterus with associated plasma cell dyscrasia. Hepatic amyloidosis usually presents with minimal clinical symptoms but portends an overall poor prognosis. Acute liver failure associated with AL amyloidosis, such as that experienced by this patient, remains exceedingly rare.
References
-
Ebert E, Nagar M (2008) Gastrointestinal Manifestations of Amyloidosis. Am J Gastroenterol 103: 776-787.
-
Petre S, Shah IA, Gilani N (2008) Gastrointestinal amyloidosis - clinical features,diagnosis and therapy. Aliment Pharmacol Ther 27: 1006-1016.
-
Park MA, Mueller PS, Kyle RA, Larson DR, Plevak MF, et al. (2003) Primary (AL) Hepatic Amyloidosis: Clinical Features and Natural History in 98 Patients. Medicine 82: 291-298.
-
Norero B, Pérez-Ayuso RM, Duarte I, Ramirez P, Soza A, et al. (2014) Portal hypertension and acute liver failure as uncommon manifestations of primary amyloidosis. Ann Hepatol 13: 142-149.
-
Lee WM, Larson AM, Stravitz T (2011) AASLD Position Paper: The Management of Acute Liver Failure: Update 2011. American Association for the Study of Liver Diseases.
-
Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, et al. (2014) International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol 15: e538-e548.
