

Clinical Medical
Reviews and Case Reports
Intravenous Immunoglobulins Efficacy in a Case of ALS with Myasthenic Symptoms
Giovanni Piccirillo1*, Francesca Trojsi1, Maria Rosaria Monsurro1, Bernardo Maria De Martino2, Francesco Habetswallner2 and Gioacchino Tedeschi1
1Metabolic and Aging Sciences, Second University of Naples, Italy
2Division of Nerophysiopathology, Hospital of National Relief, Italy
*Corresponding author: Giovanni Piccirillo, Department of Medical, Surgical, Neurological, Metabolic and Aging Sciences, Second University of Naples, 80138 Naples, Italy, Tel: +0039-0815665119, Fax: 0039-0815665095, E-mail: dr.gpiccirillo@gmail.com
Clin Med Rev Case Rep, CMRCR-2-036, (Volume 2, Issue 6), Case Report; ISSN: 2378-3656
Received: February 16, 2015 | Accepted: June 20, 2015 | Published: June 23, 2015
Citation: Piccirillo G, Trojsi F, Monsurro MR, Martino BMD, Habetswallner F, et al. (2015) Intravenous Immunoglobulins Efficacy in a Case of ALS with Myasthenic Symptoms. Clin Med Rev Case Rep 2:036. 10.23937/2378-3656/1410036
Copyright: © 2015 Piccirillo G, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Myasthenia Gravis (MG) is a neuromuscular junction disease, for which the most specific test is an increase in anti-acetylcholine receptor antibodies (anti-ACHR-Abs) titer. Myasthenic symptoms are rarely detected in patients with Amyotrophic Lateral Sclerosis (ALS). We report the case of a patient with sporadic ALS who presented at onset clinical and instrumental features suggestive of a disorder of the neuromuscular junction. She was treated with intravenous immunoglobulins for two years, with sensitive improvement in strength and delay of disease progression. The efficacy of such immunomodulating treatment in our unusual case leads to rethink the role of immune response in ALS.
Keywords
Amyotrophic lateral sclerosis, Myasthenia, Autoimmunity, Intravenous immunoglobulins
Introduction
Amyotrophic Lateral Sclerosis (ALS), a neurodegenerative disease with both upper and lower motor neurons involvement, rarely begins with fluctuating muscle weakness, which typically characterizes Myasthenia Gravis (MG), an autoimmune disorder of the neuromuscular junction. Specifically, Noseworthy et al. [1] described a patient with initial symptoms of myasthenia who subsequently developed a motor neuron disease.
Moreover, fluctuating ptosis and marked neuromuscular transmission failure in orbicularis oculi have been previously revealed in two bulbar-onset ALS patients who showed a normal titer of acetylcholine receptor (AChR) antibodies and did not improve on pyridostigmine or steroids [2].
We report the intriguing case of a sporadic ALS patient with myasthenic features at onset and clinical improvement in strength and disease severity after long-term immunomodulating therapies.
Case Report
A 54-year-old woman came to our observation for a fluctuating bulbar and spinal muscular weakness. Serum dosages of acetylcholine receptor (AChR) and muscle-specific kinase (Musk) antibodies were normal. Thorough laboratory investigation was normal, including creatinine kinase and thyroid function.
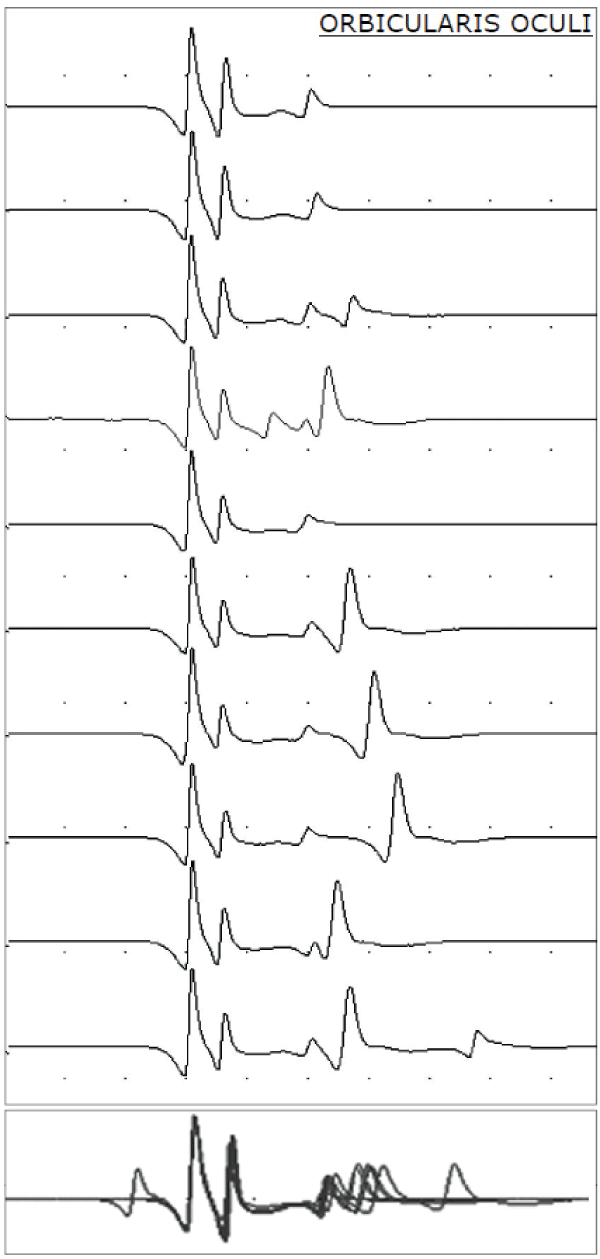
Repetitive nerve stimulation (RNS) test at 3 Hz on trapezius and orbicularis oculi muscles revealed no amplitude decrements up to 10%. A single-fiber electromyography (SF-EMG) examination in both orbicularis oculi muscles showed an abnormal jitter, measured as mean consecutive difference (57.1μs). Specifically, 9 of 20 pairs of potentials disclosed an increased jitter (>55μs) and 7 of 20 pairs of potentials disclosed blocking (Figure 1). Furthermore, in the same muscles we performed a multi-motor unit potential (MUP) analysis which showed normal values for amplitude, duration and polyphasicity, excluding neurogenic and myopathic changes. Computerized tomography of chest and brain Magnetic Resonance Imaging (MRI) was normal.
.
.
Figure 1: Single-fiber Electromyography (SF-EMG) examination of Orbicularis oculi showing abnormal jitter and potentials disclosed blocking.
View Figure 1
Considering the clinical and instrumental features indicative of a disorder of the neuromuscular junction, we began a treatment with pyridostigmine (180mg/day), prednisone (25mg/day) and azathioprine (100mg/day), obtaining a clinical improvement in fluctuations of muscular strength for one year. Afterward, we observed a generalized, progressive worsening of weakness. The patient showed a decreased strength of neck (grade 2/5 on Medical Research Council score), bulbar (2/5), arms (2/5) and legs (4/5) muscles. Thus, we treated the patient with intravenous immunoglobulins (IVIgs) (0.4g/kg/day for five consecutive days), obtaining a clinical improvement in strength (i.e., 4/5 in neck muscles, 3/5 in arms and 5/5 in legs). In particular, after the first cycle, the patient was able to eat and walk autonomously also for long distances and, even more, she was autonomous in manipulating objects and getting dressed. However, this improvement lasted about two months and, subsequently; she showed a decrease of strength in the same muscle groups involved at onset. Therefore, we repeated cyclically this treatment every two months for one year obtaining, after each IVIg course, mild improvement of strength. Six months later, we observed a progressive worsening of oropharyngeal muscles weakness associated with wasting of arms muscles and fasciculations. EMG showed pathological neurogenic motor unit changes in three sites (bulbar, upper and lower limbs). Conduction blocks were absent and sensory conductions were normal. Spinal MRI revealed no evidence of cord or root compression. Diagnostic criteria for ALS were fulfilled [3]. The patient was treated with riluzole (50mg x two/day) and high-dose IVIgs, showing clinical stability on MRC and ALS Functional Rating Scale-Revised scales six months later.
Discussion
We believe that in our ALS patient the negative results for antibodies against AChR and Musk, detected respectively in about 85% and 10% of MG patients, can evoke some doubts about the diagnosis of MG. In such cases a cell-based assay pointed towards detect antibodies to clustered ACh receptors should be done, being positive in up to 50% of "seronegative" myasthenia patients [4]. However, orbicularis oculi is not usually affected in ALS, especially in case of increased jitter without signs of denervation-reinnervation, typical of MG [2]. This raises the question whether there is an autoimmune component in some cases of ALS that may interfere with immunomodulation of neuromuscular junction, causing a motor neuronopathy with myasthenia-like features or co-occurrence of ALS with MG. To this regard, the effects of immune system in the pathogenesis of ALS have been widely studied, finding that autoimmunity can play both neuroprotective and pathogenetic roles in the nervous system of ALS patients [5]. In fact, multiple components of immune system, including T and B lymphocytes, Natural Killer cells, macrophages, mast cells and microglia, have been shown to participate in limiting toxic damage to neurons and glial cells into the neurodegerative process, promoting mechanisms of proliferation of neural progenitor stem cells, their migration in sites of injury and cellular repair [5].
Conversely, among the most intriguing pathogenetic mechanisms correlated with both immune-mediated impairment of motor neurons and injury of neuromuscular junction, it was demonstrated that serum immunoglobulins from ALS patients (ALS-IgG) may interact with neuromuscular junction, especially voltage-dependent calcium channels, in experimental models of autoimmune motor neuron disease. This may result in a cascade of increased intracellular calcium in the motor nerve terminals and increased acetylcholine release, which finally lead to motor neurons death [6]. Specifically, the absence of P/Q-type calcium channel could reduce ALS-IgG binding to mouse neuromuscular junctions and suppress antibody effects on spontaneous acetylcholine release [6]. Recently antibodies against LRP4, a membrane protein that is crucial in the development and function of motor neurons and neuromuscular junctions, have been detected not only in a fair percentage of ALS patients, seronegative for anti AChR and anti Musk, but also in some MG patients [7]; this is another evidence that even if a critical role of autoimmunity in ALS is not widely accepted, it may be a critical issue in the pathogenesis of that disease.
In particular in favour of a probable pathogenetic role of autoimmunity in our case, we found an improvement of weakness after initial implementation of immunomodulating therapies (e.g., prednisone and azathioprine) and a probable delay of disease progression after one year of high-dose therapy with IVIgs. Previously, this treatment has been found to be effective only in some motor neuron diseases associated with monoclonal gammopathy or anti-ganglioside antibodies or both, resulting to be negligible in sporadic ALS [5]. Hopefully, clinical follow-up will reveal the prognostic implications of IVIg treatment in our case, especially in slowing disease progression.
Undoubtedly, the association between ALS and MG may be exceptional, but we conjecture that sometimes it has been underestimated. Furthermore, the multiple aspects of autoimmunity in ALS are beginning to be appreciated with novel and interesting implications on potential pharmacologic targets and therapeutic approaches.
References
-
Noseworthy JH, Rae-Grant AD, Brown WF (1988) An unusual subacute progressive motor neuronopathy with myasthenia-like features. Can J Neurol Sci 15: 304-309.
-
Pinto S, de Carvalho M (2008) Amyotrophic lateral sclerosis patients and ocular ptosis. Clin Neurol Neurosurg 110: 168-170.
-
Brooks BR, Miller RG, Swash M, Munsat TL, World Federation of Neurology Research Group on Motor Neuron Diseases (2000) El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 1: 293-299.
-
Jacob S, Viegas S, Leite MI, Webster R, Cossins J, et al. (2012) Presence and pathogenic relevance of antibodies to clustered acetylcholine receptor in ocular and generalized myasthenia gravis. Arch Neurol 69: 994-1001.
-
Calvo A, Moglia C, Balma M, Chio A (2010) Involvement of immune response in the pathogenesis of amyotrophic lateral sclerosis: a therapeutic opportunity? CNS Neurol Disord-Drug Targets 9: 325-330.
-
Gonzalez LE, Kotler ML, Vattino LG, Conti E, Reisin RC, et al. (2011) Amyotrophic lateral sclerosis - immunoglobulins selectively interact with neuromuscular junctions expressing P/Q type calcium channels. J Neurochem 119: 826-838.
-
Tzartos JS, Zisimopoulou P, Rentzos M, Karandreas N, Zouvelou V, et al. (2014) LRP4 antibodies in serum and CSF from amyotrophic lateral sclerosis patients. Ann Clin Transl Neurol 1: 80-87.
