

International Journal of Neurology and Neurotherapy
Anterior Junction Syndrome Caused by Neuromyelitis Optica
Ma Eugenia García García*, Javier Casas Limón, Aida Orvíz García and Alberto Marcos Dolado
Neurology Department, Hospital Clínico San Carlos, Madrid, Spain
*Corresponding author:
Ma Eugenia García García, Neurology Department, Hospital Clínico San Carlos, C/Profesor Martín Lagos s/n 28040 Madrid, Spain, Tel: +0034637860819, Fax: +0034913303457, E-mail: mariugarciagarcia@hotmail.com
Int J Neurol Neurother, IJNN-3-045, (Volume 3, Issue 2), Case Report; ISSN: 2378-3001
Received: November 22, 2015 | Accepted: April 06, 2016 | Published: April 08, 2016
Citation: García García ME, Limón JC, García AO, Dolado AM (2016) Anterior Junction Syndrome Caused by Neuromyelitis Optica. Int J Neurol Neurother 3:045. 10.23937/2378-3001/3/2/1045
Copyright: © 2016 García García ME, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
The anterior junction syndrome is a specific manifestation caused by optic nerve involvement at the junction with the optic chiasm and the contralateral inferonasal nerve fibers (Wilbrand's Knee). Lesions at this point show a specific pattern of visual impairment which is characterized by an advanced monocular visual field loss, together with circumscribed visual field defects, respecting the vertical midline in the other eye (junctional scotoma). Although pituitary adenomas are the most frequent cause, demyelinating lesions may also be responsible.
We report a 65-year-old woman with painless loss of vision in the right eye who one month later developed temporal visual hemifield defect in the left eye. Different diagnoses were considered, however, the positivity for anti-aquaporin-4 (AQP4) antibodies was the decisive factor in the final diagnosis. The patient received intravenous methylprednisolone and plasma exchange and after several days she recovered the vision in the left eye but not in the right eye. Neuromyelitis optica (NMO) is a relapsing demyelinating disorder, with a different etiology from multiple sclerosis (MS) and with a higher morbidity and mortality. Early detection of AQP4 antibodies and institution of appropriate immunotherapy can be the key to a better prognosis.
Keywords
Anterior junction syndrome, Neuromyelitis optica, Anti-aquoporin-4 antibodies, Wilbrand's Knee
Introduction
Lesions at the optic chiasm may produce specific visual field defects that allow us to make a topographical diagnosis. Bitemporal hemianopsia (22% of the cases) and the anterior junction syndrome (13%) are the two most common visual field defects according to the study performed by Schieffer in 2004 [1].
The anterior junction syndrome is caused by a lesion located at the junction between the optic nerve and involving the optic chiasm itself, which also affects the contralateral eye inferonasal fibers, and forms Wilbrand's Knee. It is characterized by monocular advanced visual field loss together with circumscribed visual field defects respecting the vertical midline in the other eye (junctional scotoma) [2,3].
Chiasmal lesions may be categorized as intrinsic, with the lesion involving the substance of the optic chiasm itself, or extrinsic, due to mechanical compression from adjacent structures. A wide variety of possible etiologies may cause a chiasmal syndrome: neoplastic processes (such as meningiomas and gliomas) infectious causes (syphilis, tuberculosis), inflammatory lesions (sarcoidosis, demyelinating diseases), vascular (carotid aneurism) or congenital lesions such as a Rathke cleft cyst. Pituitary adenomas are, however, the most frequent causes [4-6]. Among the demyelinating disorders, it is important to make a correct differential diagnosis between MS (the most common demyelinating disorder) and NMO (or Neuromyelitis optica spectrum disorders) because, although both of them are autoimmune diseases and may have the same symptoms, the pathogenesis, the treatment and therefore the prognosis is different. The detection of serum AQP4 antibody may be the key tool to distinguish between both disorders.
Case Report
A 65-year-old woman is referred to the neurologist by the casualty department to evaluate a sudden occurrence of impaired vision in the left eye. The only data in her background was a history of arterial hypertension and a painless loss of vision in the right eye which had happened one month before.
The patient suffered this new loss of vision one day before when she wanted to read and could not see the left side of the book. She realized that when she turned her head to the left the text appeared. She did not experience pain when moving her eyes neither did she have diplopia, ptosis, headache, sensory or motor disturbances nor any other neurological disorder. She had no ocular trauma, infection or radiation history. At that time, the patient was being evaluated by an ophthalmologist for right eye vision loss. It had started 40 days before visiting the casualty department, at the bottom of the right eye's visual field, and it progressed over one week until complete amaurosis. The findings of a slit lamp biomicroscopy, intraocular pressure measurements and the ocular motility examinations were all normal. The brain magnetic resonance imaging (MRI) with gadolinium and the blood tests (including folic acid, vitamin B12, thyroid hormones, protein sedimentation rate and angiotensin converting enzyme), performed by the ophthalmologist, were normal. She was diagnosed with right ischemic optic neuropathy in the right eye and she was prescribed an antiplatelet drug.
When we saw her in consultation, her temperature was 36.5°C, her heart rate 75 beats per minute and her blood pressure 130/80 mmHg. During the visual exploration, the patient had a mydriatic, unresponsive right pupil, with a relative afferent pupillary defect. The fundoscopy showed a right papillary atrophy. Left eye visual acuity was normal; right eye showed amaurosis. The fluorescein angiography was normal and the perimetry showed a temporal visual hemifield defect in the left eye and blindness in the right eye (Figure 1a and Figure 1b). Eye movements were normal there was no ptosis or conjunctival erythema.
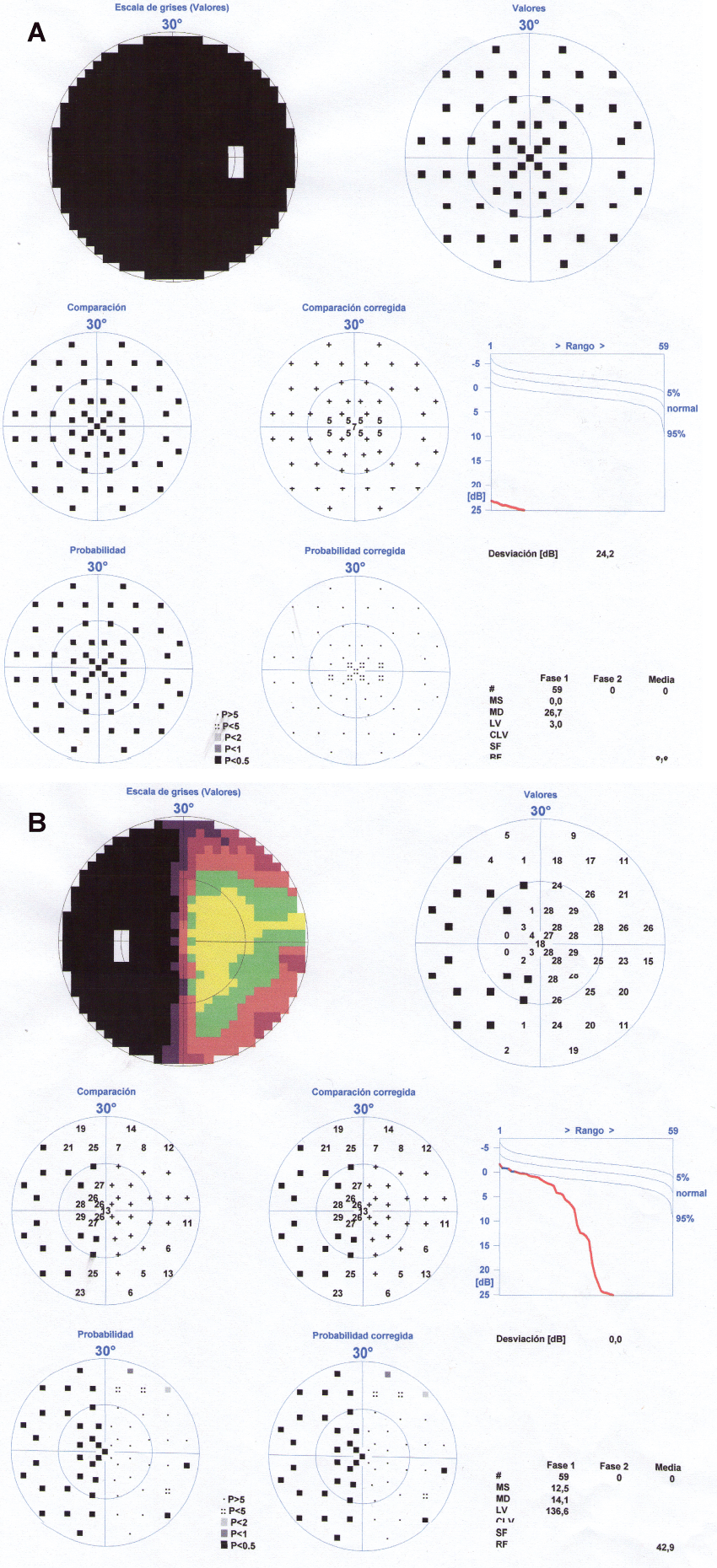
.
Figure 1(a & b): Visual perimetry showing complete blindness in her right eye and a loss of vision in the left temporal hemifield of the left eye (junctional scotoma).
View Figure 1
In order to clarify the diagnosis, the patient was admitted to the hospital. We performed immunological tests including antinuclear antibodies, antineutrophil cytoplasmic antibodies, anti-DNA antibodies as well as paraneoplastic antibodies (anti-Hu, anti-Ri, anti-Yo and anti-recoverin) which were negative. Serologic testing for HIV, syphilis, cytomegalovirus, herpes simplex virus, borrelia, cat scratch disease and cultures for common pathogens and mycobacterias were also negative.
A new brain MRI (sequence T2) showed a hyperintense lesion at the right optic nerve in its junction with optic chiasm (Figure 2a, Figure 2b and Figure 2c). The optical coherence tomography (OCT) revealed a thinner than normal retinal nerve fiber layer in the right eye. A lumbar puncture was performed extracting a clear cerebrospinal fluid (CSF) with a normal pressure. The analysis of the CSF was: glucose 94 mg/dl, proteins 32 mg/dl and one cell. Oligoclonal bands were also negative. A positron emission tomography (PET) did not show any tumor. After performing the lumbar puncture, the patient was started on empirical therapy with intravenous methylprednisolone at a dose of 1000 mg a day, for five days, followed by a descending dose of corticosteroids. One week later the patient's neurological status was unchanged so we decided to administer plasmapheresis.
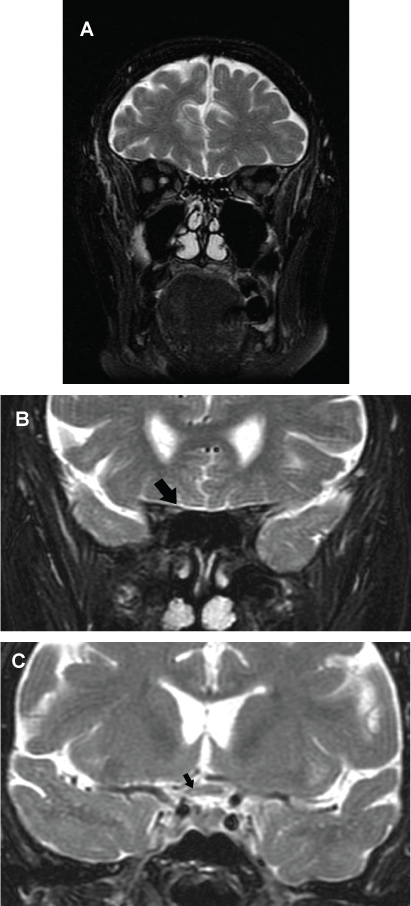
.
Figure 2: (a & b) Coronal T2-weighted brain MRI showing a right optic nerve hyperintensity (arrow); (c) Coronal T2-weighted image showing a subtle demyelinating lesion involving the optic chiasm (arrow).
View Figure 2
The patient was discharged and in the subsequent review, a week later, she reported a significant improvement in the visual field of her left eye with a complete recovery one month later (Figure 3a and Figure 3b). The right eye did not change. At this time we received the results of the AQP-4 antibodies which were positive and immunosuppressive treatment was initiated (azathioprine at a dose of 150 mg/day) together with prednisone (40 mg/day).
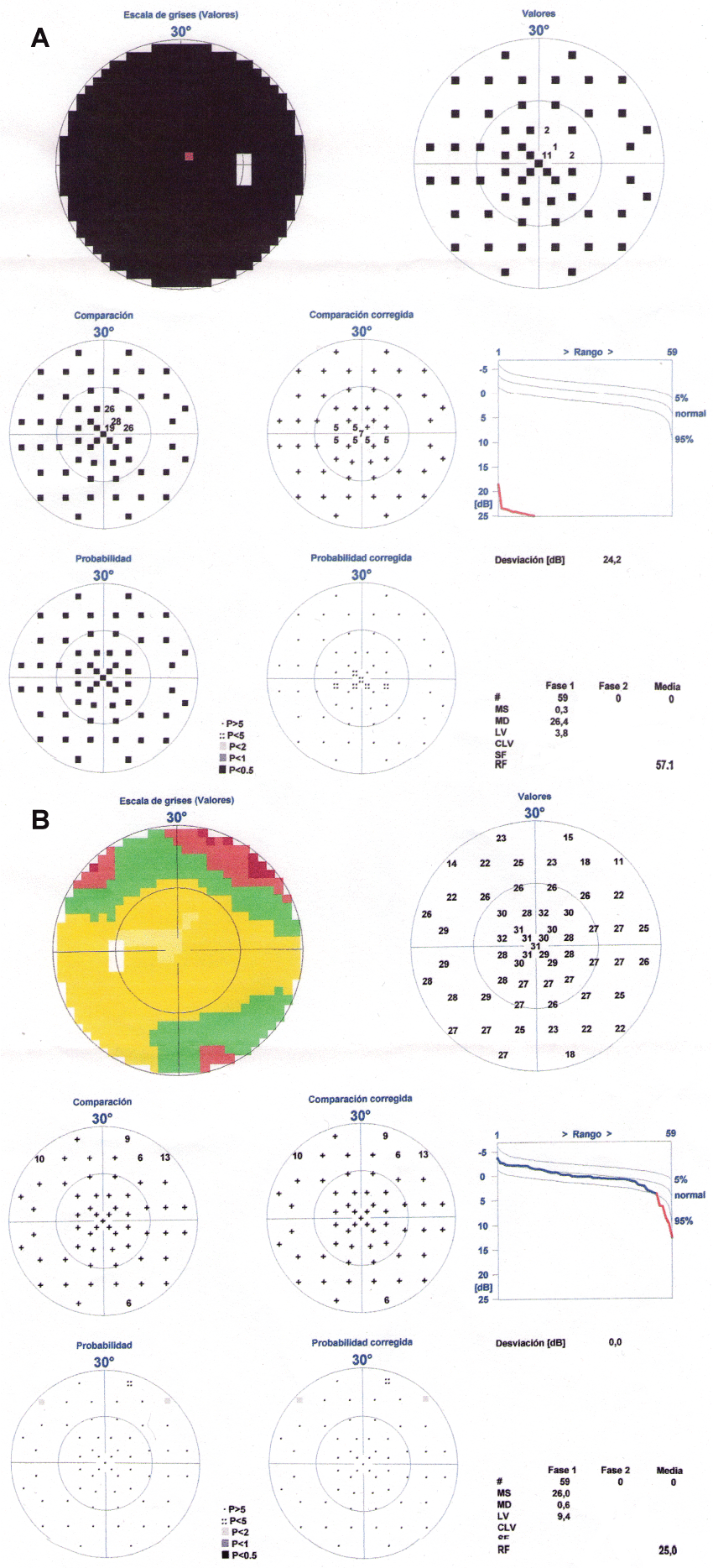
.
Figure 3(a & b): Visual perimetry after the plasma exchange. There was a resolution of the temporal visual field defect in the left eye while the right eye remained without changes.
View Figure 3
Discussion
We report the case of a healthy 65-year-old female who suffered from a loss of vision in the right eye and in the temporal visual hemifield of the left eye (anterior junction syndrome) due to NMO spectrum disorders (NMOSD). This is considered to be an autoimmune, antibody-mediated disease especially since the identification of a specific autoantibody (NMO-IgG) directed against the water channel protein, AQP4, which is mainly expressed in astrocytes foot processes at the blood brain barrier.
NMOSD affect all races and ages, but has a clear and notable female preponderance. Traditionally, they were characterized by severe attacks of optic neuritis and myelitis, either simultaneously or in isolation. Optic neuritis (ON) is the initial manifestation of NMO in 77% of patients and it has a worse prognosis than MS (only 43% of the patients will completely recover while 30% will remain blind after the first attack). The relapses are also more frequent and earlier than in MS [7-10]. New international consensus criteria for the diagnosis of NMOSD have been established by Wingerchuck et al. (2015). Certain clinical presentations such as bilateral optic nerve involvement and posterior nerve predominance (especially with extension into the optic chiasm) are considered suggestive of NMOSD [11].
Some authors have hypothesized that the differing pathogenic mechanisms of NMOSD and MS may result in different patterns of visual field defects as findings of ON. They discovered that patients with NMOSD had a higher incidence of non-central scotoma and the altitudinal hemianopia was the most frequent. This visual field defect is highly characteristic of ischemic optic neuropathy, therefore they suggested the possibility of an ischemic mechanism mediated by AQP4 [7].
Due to the prognosis and treatment being different, the distinction between NMOSD and MS is crucial. Medical history, brain MRI, the presence or not of oligoclonal bands as well as the assessment of AQP4 antibodies are the tools used to distinguish the two entities [11,12]. In our case, the cerebral MRI showed a demyelinating lesion in the right optic nerve with extension into the optic chiasm without any other demyelinating lesions. A study performed by Khana et al. (2013) showed no statistically significant differences between the presence, degree or type of signal alteration and contrast enhancement of the affected nerve segments between the NMO and MS groups [13]. However the new criteria for NMOSD consider lesions involving the posterior aspects of the optic nerve or the chiasm more associated with this disorder. Findings from the OCT could potentially differentiate them because NMO usually shows a thinner retinal nerve fiber layer than MS which suggests a greater axonal injury [14].
As NMO spectrum disorders are rare entities, there is no available evidence from randomised controlled treatment trials for acute relapses or for their prevention and the recommendations are based only on case reports or small series of cases.
Due to the severity of the NMOSD attacks and the high risk of disability, treat¬ment should be instituted as soon as possible. The treatment has two main objectives: one is to con¬trol the inflammatory damage in acute attacks and the other one is to avoid relapses. The former is based on high dose intravenous methylprednisolone (1000 mg a day for five days) and plas¬mapheresis. Early initiation of plasma exchange (PE), within 20 days of an attack's onset, predicts greater likelihood of a clinical response. In contrast to evidence of a positive effect of PE, the efficacy of intravenous immunoglobulins has not been demonstrated in patients with severe attacks [15].
The relapse prevention is based on early and maintenance immunosuppressive treatments which clearly and dramatically reduce relapses, and can lead up to a six-fold relapse reduction compared with pretreatment [16]. Considering the antibody-driven hypothesis, treatment should target B cells. For this reason, mycophenolate mofetil, azathioprine, mitoxantrone and monoclonal antibodies (rituximab) have been used in different cases [17].
Sero-negative patients should be treated in the same way as sero-positive patients if they fulfil the diagnostic criteria for NMOSD.
Conclusion
Demyelinating lesions in the chiasm are uncommon and they are manifested with visual field defects as the anterior junction syndrome. One of the etiologies can be neuromyelitis optica spectrum disorders. NMO antibody plays an important role in aiding the diagnosis.
References
-
Schiefer U, Isbert M, Mikolaschek E, Mildenberger I, Krapp E, et al. (2004) Distribution of scotoma pattern related to chiasmal lesions with special reference to anterior junction syndrome. Graefes Arch Clin Exp Ophthalmol 242: 468-477.
-
Hoyt WF (1970) Correlative functional anatomy of the optic chiasm. 1969. Clin Neurosurg 17: 189-208.
-
Shin WJ, Song BJ, Kim JM (2002) Junctional scotoma in giant cerebral aneurysm. Korean J Ophthalmol 16: 124-129.
-
Kupersmith MJ, Straga J, Zeiffer B, Kraker R (2001) Junctional scotoma in acute optic neuritis or inflammation of the ‘knee'. Invest Ophthalmol Vis Sci 42 [Suppl]: 326.
-
Foroozan R (2003) Chiasmal syndromes. Curr Opin Ophthalmol 14: 325-331.
-
Mejico LJ, Miller NR, Dong LM (2004) Clinical features associated with lesions other than pituitary adenoma in patients with an optic chiasmal syndrome. Am J Ophthalmol 137: 908-913.
-
Nakajima H, Hosokawa T, Sugino M, Kimura F, Sugasawa J, et al. (2010) Visual field defects of optic neuritis in neuromyelitis optica compared with multiple sclerosis. BMC Neurol 10: 45.
-
Chiquete E, Navarro-Bonnet J, Ayala-Armas R, Gutiérrez-Gutiérrez N, Solórzano-Meléndez A, et al. (2010) Neuromielitis óptica: actualización clínica. Rev Neurol 51: 289-294.
-
Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG (1999) The clinical course of neuromyelitis optica (Devic's syndrome). Neurology 53: 1107-1114.
-
Merle H, Olindo S, Bonnan M, Donnio A, Richer R, et al. (2007) Natural history of the visual impairment of relapsing neuromyelitis optica. Ophthalmology 114: 810-815.
-
Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, et al. (2015) (International Panel for NMO Diagnosis) International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 85: 177-189.
-
Filippi M, Rocca MA (2004) MR imaging of Devic's neuromyelitis optica. Neurol Sci 25 Suppl 4: S371-373.
-
Khanna S, Sharma A, Huecker J, Gordon M, Naismith RT, et al. (2012) Magnetic resonance imaging of optic neuritis in patients with neuromyelitis optica versus multiple sclerosis. J Neuroophthalmol 32: 216-220.
-
Ratchford JN, Quigg ME, Conger A, Frohman T, Frohman E, et al. (2009) Optical coherence tomography helps differentiate neuromyelitis optica and MS optic neuropathies. Neurology 73: 302-308.
-
Collongues N, de Seze J (2011) Current and future treatment approaches for neuromyelitis optica. Ther Adv Neurol Disord 4: 111-121.
-
Palace J, Leite MI, Jacob A (2012) A practical guide to the treatment of neuromyelitis optica. Pract Neurol 12: 209-214.
-
Awad A, Stüve O (2011) Idiopathic transverse myelitis and neuromyelitis optica: clinical profiles, pathophysiology and therapeutic choices. Curr Neuropharmacol 9: 417-428.
