

International Journal of Ophthalmology and Clinical Research
Novel Deletion in the CNNM4 Gene in Siblings with Jalili Syndrome
F Kiessling1*, D Mitter1, T Langmann2, D Müller1 and H Tegetmeyer3
1Institut für Humangenetik, Universitätsklinikum Leipzig, Germany
2Zentrum für Augenheilkunde, Universitätsklinikum Köln, Germany
3Klinik und Poliklinik für Augenheilkunde, Universitätsklinikum Leipzig AöR, Germany
*Corresponding author: Franziska Kiessling, Institut für Humangenetik, Universitätsklinikum Leipzig, Philipp-Rosenthalstr 55, 04103 Leipzig, Germany, Tel: +40 341 97 23800, Fax: +49 341 97 23819, E-mail: franziska.kiessling@medizin.unileipzig.de
Int J Ophthalmol Clin Res, IJOCR-3-046, (Volume 3, Issue 1), Research Article; ISSN: 2378-346X
Received: October 30, 2015 | Accepted: January 14, 2016 | Published: January 16, 2016
Citation: Kiessling F, Mitter D, Langmann T, Müller D Tegetmeyer H (2016) Novel Deletion in the CNNM4 Gene in Siblings with Jalili Syndrome. Int J Ophthalmol Clin Res 3:046. 10.23937/2378-346X/1410046
Copyright: © 2016 Kiessling F, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Different mutations in the CNNM4 (OMIM 607805) gene are known to cause Jalili syndrome (OMIM 217080) which is characterized by a combination of cone-rod dystrophy and amelogenesis imperfecta. In particular one homozygous missense mutation Leu438Profs*9 in exon 1 of the CNNM4 gene has been described in patients originating from the Kosovo. This mutation causes a frameshift and generates a new stop codon in the same exon. The two patients described here carry the same Leu438Profs*9 mutation in a heterozygous state. In addition they also carry a novel heterozygous deletion which has not been described in the literature. The deletion includes 29 base pairs and is also located in exon 1 of the CNNM4 gene. A genetic analysis of the parents revealed both mutations to be compound heterozygous and are therefore the likely cause of Jalili syndrome in the family.
Keywords
Jalili syndrome, CNNM4, Leu438Profs*9, 29 basepair deletion, Cone-rod dystrophy, Amelogenesis imperfect
Introduction
Cone-rod dystrophy (CRD) in combination with amelogenesis imperfecta (AI) is a characteristic pattern in a new syndrome first described in 1988 by Jalili & Smith [1] in an extended Arab family from the Gaza Strip with family members affected with photophobia, nystagmus, achromatopsia and abnormal, discolored teeth. Since then additional families affected both with CRD and AI have been described with Jalili syndrome [2-4]. Cone-rod dystrophies are a heterogeneous group of genetically related retinal diseases characterized by progressive loss of visual acuity and achromatopsia [5]. Photophobia and often night-blindness the patients are unable to differentiate colors. Amelogenesis imperfecta includes several different clinical features such as hypoplastic, hypomature or hypocalcified enamel of the teeth [6]. Mutations in the CNNM4 gene are causative for Jalili syndrome [2,7,8]. The CNNM4 gene, located on chromosome 2q11, spans 51kb of genomic DNA in seven exons [9]. It encodes a magnesium transport protein which is necessary for many cellular functions, including a role as cofactor for enzymes of the phototransduction cascade [7,10].
The description of an affected sib pair from Kosovo, affected both with CRD and AI, suggests that the combination of this phenotype may exist as a genetically homogeneous syndrome [2]. The reported sibs were genetically analyzed and a homozygous mutation in exon 1 of the CNNM4 gene, further in this report termed the Kosovo mutation, has been ascertained. We here report a family with sibs clinically affected with Jalili syndrome carrying the Kosovo mutation in a compound heterozygous state. This mutation occurred with an allelic previously unknown deletion (c.694_722del) located in exon 1 of the CNNM4 gene.
Patients and Methods
Patients
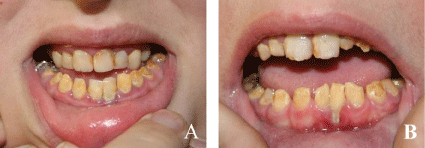
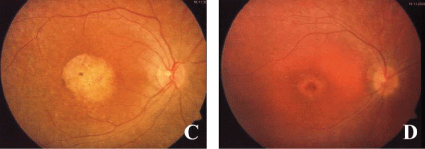
The family members described here originate from Kosovo. The affected sibs are the only children of non-consanguineous parents. Because of ambiguous retinal findings a genetic diagnostic was taken into consideration. Because of both, the kosovan ancestry and the clinical signs a genetic cause for Jalili syndrome was suspected. At the time of examination and molecular diagnosis the index patient (female) was 20 years old, her brother was 16 years old. The anamnesis resulted in both with loss of visual acuity at the age of about four years. Also at the age of about four years conspicuous dental findings were described. The deciduous teeth of both sibs and the permanent teeth showed yellowish discoloration and carious changes with enamel defect, typical signs of amelogenesis imperfecta (Figure 1). At the age of six years an ophthalmologic examination of the index patient revealed rod-cone dystrophy with juvenile macular degeneration (Figure 2) based on the following findings: fixation nystagmus, range of vision with concentrical constriction of approximately 30 percent, vision defects in the dark. The Panel-D15 test revealed bilateral multiple deviations along the tritan axis. At the age of ten years a further ophthalmological examination confirmed these clinical results and an electroretinography (ERG) revealed a maximal response scotopic with a reduced amplitude. A dysfunction including cones as well as rods was diagnosed. For the index patient the achromatopsia was diagnosed at the age of fifteen by one of the authors (D M) at the Hospital for Pediatrics and Adolescent Medicine in Chemnitz, Germany. At the age of 17 years a control examination revealed a progression of the clinical signs such as retinal pigment degeneration, bilateral effaced ERG-response for scotopic and photopic range, restriction of visual field to 10 degree of the right eye and 12 degree diameter (vertical) and 18 degree diameter horizontal of the left eye. Panel- D15 test again showed bilateral multiple deviations along the tritan- and the deutan axis. Altogether several signs, which are typical for Jalili syndrome, were present at the index patient and her brother. The parents do not have any problems with vision and have no dental changes. All participating persons gave their informed consent prior to the genetic examination.
.
Figure 1: Teeth showing amelogenesis imperfecta and the caries in the sister at the age of fifteen (A) and the brother at the age of eleven (B).
View Figure 1
.
Figure 2: Fundus of the patients: (C) macular dystrophy with pigmentary degeneration at the sisters eye, (D) Bull's eye maculopathy at the brothers eye.
View Figure 2
Methods
Blood samples were taken after informed written consent. Genomic DNA was purified using the MasterPure™ DNA Purification Kit for Blood Version II (Epicentre Technologies Corp., Chicago). In view of the large size of exon 1 (1500 bp), it was divided into five overlapping fragments (a-e) (Table 1). The Primers were designed using the 'Primer3' software (http://frodo.wi.mit.edu/). For the GC-rich sequence in parts a and b of exon 1 the Advantage®-GC Genomic LA Polymerase Mix (Clontech Laboratories Inc., USA) was used for amplification according to the manufacturer's protocol. The PCR reaction was performed with the following modifications: initial denaturation at 95°C for 10 minutes, followed by 35 cycles for denaturation at 95°C for 1.5 minutes, 1-minute hold at the indicated annealing temperature (Table 1), and extension at 72°C for 3 minutes, with a final extension step at 72°C for 10 minutes. For amplification of the fragments c-e of exon 1 a standard PCR (Maxima Hot Start Taq DNA Polymerase, Thermo Fisher Scientific Inc. Fermentas, PA/USA) was used according to the manufacturer's protocol. The PCR conditions were as follows: initial denaturation at 95°C for 7 minutes, followed by 35 thermo cycles for denaturation at 95°C for 30 seconds, 1-minute hold at the indicated annealing temperature (Table 1), and extension at 72°C for 1 minute, with a final extension step at 72°C for 10 minutes. Samples were sequenced using the Big Dye Terminator v3.1 Cycle Sequencing Reaction kit (Applied Biosystems, Foster City, CA/USA) according to the manufacturer's protocol. For analysis the 3500 Genetic Analyzer (Applied Biosystems, Foster City, CA) was used. Bidirectional sequencing using primers specific for CNNM4 exon 1 were used to screen for the expected mutations (Table 1). Sequence variants were analyzed using CodonCode Aligner Vs. 3.7.1, mutation data were indicated using the reference-sequence: NM_020184.3.
![]()
Table 1: Primer Sequences for the fragments of CNNM4 Exon 1.
View Table 1
Results
The sequencing analysis of the index patient revealed the presence of the known c.1312dupC mutation in exon 1 located in the CBS domain of the CNNM4 gene, the so called Kosovo mutation. The duplication of cytosine leads to a substitution of the amino acid residue at amino acid position 438 and the resulting frameshift generates a stop codon nine amino acid residues downstream (p.Leu438Profs*9). Presumably this generates a truncated non-functional protein. Surprisingly this mutation in the index patient was present in a heterozygous state. A second heterozygous mutation was identified in fragment 1c (c.694_722del), which was upstream of the known mutation. The heterozygous deletion of 29 base pairs is leading to a substitution of isoleucine to proline at amino acid position 232. Since this frameshift generates a stop codon 80 amino acid residues downstream (p.Ile232Profs*80) it will lose its function as in the typical Kosovo mutation. The analysis of the remaining exons of the CNNM4 gene did not reveal any abnormalities. Based on the clinical phenotype of the patients, we assume both mutations to be compound heterozygous and thus to be causative for the Jalili-syndrome. The analysis by direct sequencing revealed that the affected brother also carry both mutations in a compound heterozygous state. At father's DNA sample the so-called Kosovo mutation was detectable in a heterozygous state. The mother carries the mutation in exon 1, fragment 1c of the CNNM4 gene in a heterozygous state. The remaining exons of the CNNM4 gene did not contain any mutations in the index patient. The brother as well as the parents was tested for exon 1 of the CNNM4 gene only.
Discussion
CNNM4 encodes a protein which is suggested to have functions in magnesium transport. [11] Studies revealed that CNNM4 mediates transcellular Mg2+ transport and posseses characteristics of a Na+/Mg2+ exchanger [10]. Magnesium is an essential element that is required for the catalytic activity of numerous met alloenzymes [12]. Deficiency of Mg2+ among others can cause several cardiovascular, neurological, and metabolic diseases [13]. The loss of functional CNNM4 protein observed here may have pathological effects such as hypoplastic and hypomineralized enamel resulting in amelogenesis imperfecta present in families clinically diagnosed with Jalili syndrome [2].
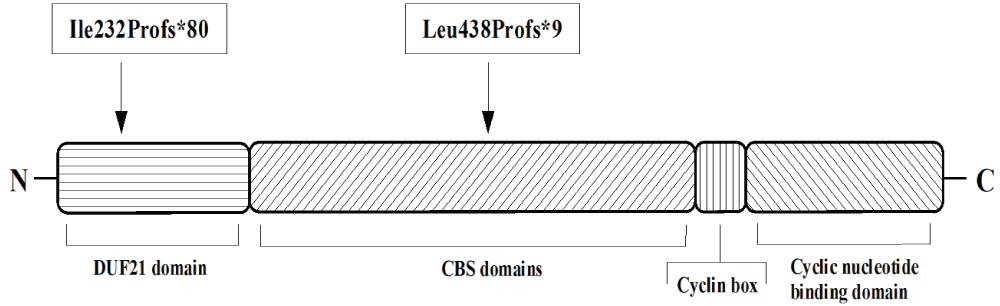
The CNNM4 protein consists of four functional domains (Figure 3), whereas the DUF21 domain contains four transmembrane helices (TM1-4) [12]. The novel deletion of 29 bp found in our study involves the amino acid residue 232, located between TM1 and TM2. Furthermore, the Kosovo mutation c.1312dupC affects the CBS-domain of the protein. Both are located in exon 1, the promoter region of the CNNM4 gene. Thus an alteration of the highly conserved sequences very likely leads to a loss of protein. Additional investigations using RNA- and protein analyses could reveal further details. The typical phenotype of patients affected with Jalili syndrome has been classified into two types, A and B, by Jalili [14]. In type A ocular features occur in early infancy and can be present at birth, whereas the type B phenotype is milder and of later onset [14]. The macular degeneration together with the loss of visual acuity at early infancy of both sibs indicate the presence of type A.
.
Figure 3: Structure of CNNM4 including protein domains (according to Parry et al. [7]) and mutations discovered in the members of the family described in this study. Abbreviations: CBS domain: cystathionine beta-synthase core domain; DUF21 domain: domain of unknown function DUF21 [1].
View Figure 3
Based on the Kosovan ancestry of our patients, we expected to find the Kosovo mutation Leu438Profs*9 which has been described previously in different patients in a homozygous state [2,7,15-17]. Our findings reveal that the Kosovo mutation may be associated with a different previously unknown mutation in the CNNM4 gene as genetic cause for the Jalili-syndrome. Thus our findings extend the spectrum of the mutations in the CNNM4 gene causative for Jalili syndrome. Genetic tests when suspecting Jalili syndrome should include the entire coding regions of CNNM4 including the promoter region because causative mutations are potentially located here.
Now the phenotype described includes amelogenesis imperfecta and cone-rod dystrophy as described in five affected members originating from Kosovo [2,4,7,10]. Different types of amelogenesis imperfecta exist which differ in pattern of inheritance, autosomal dominant (OMIM 104510), autosomal recessive (OMIM 204650) and X-linked (OMIM 301200) types. Autosomal recessive amelogenesis imperfecta types are more common in the Middle East [18,19]. The pathogenic mutation Leu438Profs*9 in exon 1 of the CNNM4 gene is known to be associated with Jalili-syndrome in a homozygous state. It is well known that recessive mutations can occur in religious or geographical isolates as a result of a founder effect. Patients with Jalili syndrome originate from different territories but only patients originating from the Kosovo have been found to carry the mutation Leu438Profs*9 [4,7,17,20-22]. Referring to the two cases described by Parry [2009], the clinical Jalili syndrome of the patients in this study is also caused by two compound-heterozygote mutations in the CNNM4 gene (Table 2) [7]. Considering that the family originates for at least three generations from Kosovo and thus we supported the existence of a founder effect of this mutation.
![]()
Table 2: Families with compound-heterozygote mutations in the CNNM4 gene.
View Table 2
Acknowledgments
We wish to thank the Jalili syndrome family for their participation and cooperation. We thank Dr. Scholbach for referral of the patients, and Professor Eberhard Passarge for reading the manuscript and helpful suggestions.
References
-
Jalili IK, Smith NJ (1988) A progressive cone-rod dystrophy and amelogenesis imperfecta: a new syndrome. J Med Genet 25: 738-740.
-
Polok B, Escher P, Ambresin A, Chouery E, Bolay S, et al. (2009) Mutations in CNNM4 cause recessive cone-rod dystrophy with amelogenesis imperfecta. Am J Hum Genet 84: 259-265.
-
Downey LM, Keen TJ, Jalili IK, McHale J, Aldred MJ, et al. (2002) Identification of a locus on chromosome 2q11 at which recessive amelogenesis imperfecta and cone-rod dystrophy cosegregate. Eur J Hum Genet 10: 865-869.
-
Michaelides M, Bloch-Zupan A, Holder GE, Hunt DM, Moore AT (2004) An autosomal recessive cone-rod dystrophy associated with amelogenesis imperfecta. J Med Genet 41: 468-473.
-
Huang L, Xiao X, Li S, Jia X, Wang P, et al. (2012) CRX variants in cone-rod dystrophy and mutation overview. Biochem Biophys Res Commun 426: 498-503.
-
Witkop CJ Jr (1988) Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited: problems in classification. J Oral Pathol 17: 547-553.
-
Parry DA, Mighell AJ, El-Sayed W, Shore RC, Jalili IK, et al. (2009) Mutations in CNNM4 cause Jalili syndrome, consisting of autosomal-recessive cone-rod dystrophy and amelogenesis imperfecta. Am J Hum Genet 84: 266-273.
-
Coppieters F, Van Schil K, Bauwens M, Verdin H, De Jaegher A, et al. (2014) Identity-by-descent-guided mutation analysis and exome sequencing in consanguineous families reveals unusual clinical and molecular findings in retinal dystrophy. Genet Med 16: 671-680.
-
National Center for Biotechnology Information (NCBI) (2010) Genetic Testing Registry (GTR).
-
Yamazaki D, Funato Y, Miura J, Sato S, Toyosawa S, et al. (2013) Basolateral Mg2+ extrusion via CNNM4 mediates transcellular Mg2+ transport across epithelia: a mouse model. PLoS Genet 9: e1003983.
-
Wang CY, Shi JD, Yang P, Kumar PG, Li QZ, et al. (2003) Molecular cloning and characterization of a novel gene family of four ancient conserved domain proteins (ACDP). Gene 306: 37-44.
-
Gómez García I, Oyenarte I, Martínez-Cruz LA (2011) Purification, crystallization and preliminary crystallographic analysis of the CBS pair of the human met al transporter CNNM4. Acta Crystallogr Sect F Struct Biol Cryst Commun 67: 349-353.
-
Kikuchi K, Tanaka H, Gima M, Kashiwagi Y, Shida H, et al. (2012) Abnormalities of magnesium (Mg) metabolism and therapeutic significance of Mg administration in patients with metabolic syndrome, type 2 diabetes, heart failure and chronic hemodialysis. Clin Calcium 22: 1217-1226.
-
Jalili IK (2010) Cone-rod dystrophy and amelogenesis imperfecta (Jalili syndrome): phenotypes and environs. Eye (Lond) 24: 1659-1668.
-
Zobor D, Kaufmann DH, Weckerle P, Sauer A, Wissinger B, et al. (2012) Cone-rod dystrophy associated with amelogenesis imperfecta in a child with neurofibromatosis type 1. Ophthalmic Genet 33: 34-38.
-
Luder HU, Gerth-Kahlert C, Ostertag-Benzinger S, Schorderet DF (2013) Dental phenotype in Jalili syndrome due to a c.1312 dupC homozygous mutation in the CNNM4 gene. PLoS One 8: e78529.
-
Gerth-Kahlert C, Seebauer B, Dold S, Hanson JV, Wildberger H, et al. (2015) Intra-familial phenotype variability in patients with Jalili syndrome. Eye (Lond) 29: 712-716.
-
Chosack A, Eindelman E, Wisotski I, Cohen T (1979) Amelogenesis imperfecta among Israeli Jews and the description of a new type of local hypoplastic autosomal recessive amelogenesis imperfecta. Oral Surg Oral Med Oral Pathol 47: 148-156.
-
Nusier M, Yassin O, Hart TC, Samimi A, Wright JT (2004) Phenotypic diversity and revision of the nomenclature for autosomal recessive amelogenesis imperfecta. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 97: 220-230.
-
Doucette L, Green J, Black C, Schwartzentruber J, Johnson GJ, et al. (2013) Molecular genetics of achromatopsia in Newfoundland reveal genetic heterogeneity, founder effects and the first cases of Jalili syndrome in North America. Ophthalmic Genet 34: 119-129.
-
Purwar P, Sareen S, Bhartiya K, Sayed Inayatullah SR, Bansal M, et al. (2015) Jalili syndrome presenting with situs inversus totalis and keratoconus: the first case in the Indian subcontinent. Oral Surg Oral Med Oral Pathol Oral Radiol 120: e210-e218.
-
Prasad MK, Geoffroy V, Vicaire S, Jost B, Dumas M, et al. (2015) A targeted next-generation sequencing assay for the molecular diagnosis of genetic disorders with orodental involvement. J Med Genet.s
