

International Journal of Pathology and Clinical Research
Research Progress of STK33 in Cancer Biology
Chen Chen1,2 and Jianfeng Li1,2,3*
1Department of Otolaryngology-Head and Neck Surgery, Provincial Hospital Affiliated to Shandong University, Jinan 250021, P.R. China
2Department of Pathology, Medical College, Shandong University, Jinan, 250021, P.R. China
3Shandong Institute of Otolaryngology, Jinan 250021, P.R. China
*Corresponding author:
Jianfeng li, Department of Otolaryngology-Head and Neck Surgery, Provincial Hospital Affiliated to Shandong University, Jinan 250021, P.R. China, E-mail: lijianfeng@hotmail.com
Int J Pathol Clin Res, IJPCR-2-028, (Volume 2, Issue 1), Mini Review; ISSN: 2469-5807
Received: November 24, 2015 | Accepted: February 26, 2016 | Published: February 29, 2016
Citation: Chen C, Li J (2016) Research Progress of STK33 in Cancer Biology. Int J Pathol Clin Res 2:028. 10.23937/2469-5807/1510028
Copyright: © 2016 Chen C, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
In recent years, serine/threonine kinase 33 (STK33) has attracted considerable attention in tumor biology. STK33 displays a heterogenous expression pattern, and in most tissues, expression level is low. It belongs to the CAMK family, and differentiates itself from other members of the CAMK family due to its expression pattern. STK33 is likely to participate in the dynamic changes of intermediate filament cytoskeleton depolymerization by phosphorylating vimentin, and thereby influencing the cell structure and a series of important functions. In the process of cell signal transduction, especially its interaction with certain oncogenes, STK33 plays a special role in the regulation of tumor proliferation. Of note, STK33 is involved in the “synthetic lethal” process of a variety of tumor cells, which depends on the Ras oncogene. However, studies also demonstrate that STK33 activity is nonessential in KRAS-dependent cancer cells. Overall, available data have shown that STK33, as a new target, might be of great importance in the molecular-targeted therapy for cancer.
Keyword
STK33, Molecular targeted therapy, Tumor
Introduction
Protein kinases, a protein super-family, have been extensively explored in biomedical field. As a member of this super-family, the function of Serine/threonine kinase is to phosphorylate serine and threonine residues on target proteins. This plays an important role in many physiological processes, such as DNA replication, signal transducing pathway, cell proliferation, differentiation, death, and tumorigenesis [1].
In recent years, serine/threonine kinase 33 (STK33) has attracted considerable attention in biomedicine. Initial studies show that STK33 regulates the signal transduction pathway in K-ras-dependent cancer cells and plays a special role in tumor proliferation [2]. Although a few articles about STK33 appear in literature, the studies of STK33 have become a hot topic in biology, especially, in oncology. Preliminary research suggests that STK33 may be a novel target for cancer molecular-targeted therapy [3,4].
Structure of STK33
The human STK33 gene, which is located in the human chromosome 11 region 11p15.3, contains 12 exons. The open reading frame of STK33 adds up to 1545 bp and codes for a protein of 57.8 kDa, which is subsequently confirmed to be a novel Serine/threonine kinase [4]. STK33 not only has all features of the active kinase (including the serine/threonine kinase active site and the ATP binding domain), but also shows significant similarity to Ca2+/calmodulin dependent kinase (CAMK) in structure. Therefore, STK33 is classified as the member of the CAMK family. The serine/threonine kinase family of CAMK regulates many of the biological responses observed in cells by phosphorylation [5,6].
The Distribution and the Localization of STK33
In order to explore the distribution and the localization of STK33, the expression of STK33 in different normal adult and fetal tissues was analyzed at the RNA and protein levels. The results show that STK33 displays a heterogenous and, in most tissues, low level of expression. In testis, particularly in the cells from the spermatogenic epithelia, the expression level of STK33 is significantly high. Furthermore, high expression of STK33 is detected in lung epithelia, alveolar macrophages, horizontal cells in the retina and the heart, brain and spinal cord of embryonic organs [7]. Though CAMK II is widely expressed in the central nervous system (CNS), STK33 lacks expression in the mature nerve tissue [8,9]. STK33 is mainly localized in the cytoplasm, which reflects the characteristics of most soluble proteins. Consequently, STK33 exhibits a tissue-specific expression and is closely related to different stages of development in various organs. Obviously, there are some similarities and differences between STK33 and the subtype members of CAMK family in expression patterns. Both in vitro and in vivo experiments show that STK33 needs Ca2+/calmodulin to be activated. STK33 canautophosphorylate serine and threonine, but, whether the autophosphorylation is a prerequisite for the function of STK33 has yet to be fully resolved [7].
It has been demonstrated that STK33 and vimentin coexist in different tissues and cells, and that STK33 is able to specifically phosphorylate the non-α-helical amino-terminal domain of vimentin in vitro [10,11]. Furthermore, co-immunoprecipitation experiments with the cultured cell extracts reveal that STK33 and vimentin are closely associated in vitro [10,11]. Vimentin, an important type III intermediate filament protein, is mainly located in the embryonal tissue and the adult cells, which arise from mesenchymal tissues. The expression of vimentin in tumor cells is related to the growth and invasion of the tumor. Intermediate filament protein is an important structure in eukaryotic cells, which makes up the cytoskeleton together with microtubule and actin microfilament. In the process of cell movement, mitosis or cell division and tumorigenesis, dynamic changes of intermediate filament protein combination are especially apparent. In this regard, it can be speculated that STK33 is likely to participate in the dynamic changes of intermediate filament cytoskeleton depolymerization by specifically phosphorylating vimentin, thus influencing the cell structure and a series of important functions. In this way, STK33 might take part in the initiation and development of tumor.
STK33-Oncogene
The reason that STK33 has been attracted so much attention because it is recognized to be a novel gene, which participates in "synthetic lethality" of various Ras-dependent tumor cell lines. Synthetic lethality means the mutation in two or more genes can lead to death of cell. Namely, the mutation of a single gene will not cause cell death, whereas, mutation in two or more genes can result in death of cells. The significance of screening synthetic lethal genes is to find the new drug target, so as to kill the cancer cells without damaging the normal ones [12]. So far, the mechanism of synthetic lethal is still unclear. While the use of synthetic lethality has been around for decades in model organism studies, it is only recently that it has been applied to cancer therapy with the development of RNA interfering technology [13].
Scholl et al. [14] use high-throughput RNA interference to identify synthetic lethal interactions in cancer cells harboring mutant K-ras gene. They found that cells which are dependent on mutant K-ras exhibit sensitivity to suppression of STK33 regardless of tissue origin. This means that knockdown or mutation of STK33 may inhibit the ability of proliferation in cancer cells, and the ability of tumorigenesis in immunodeficient mice. Nevertheless, STK33 is not required by KRAS-independent cells. The same phenomenon is shown in many K-ras dependent cancers, such as AML, multiple myeloma, breast cancer, colon cancer, pancreatic cancer, lung cancer, glioblastoma, prostate cancer, and T-ALL [14]. This indicates that both mutant K-ras and STK33 are essential components of the signaling pathways to maintain the transformed phenotype of cancer cells. The fact that they cannot substitute for each other reflects the characteristic of the mutant K-ras-driven cancer cells to be dependent on the persistent expression of both genes (Figure 1).
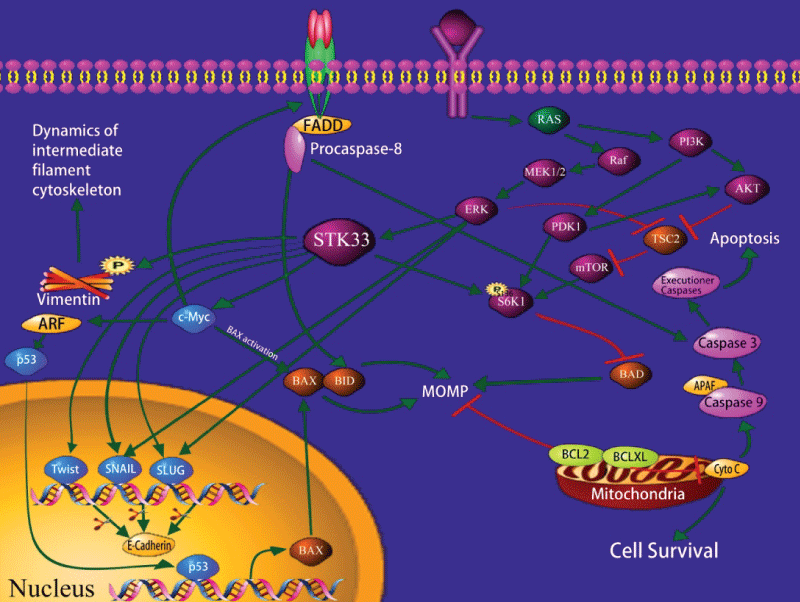
.
Figure 1: Schematic representation of the intracellular pathways relevant to STK33.
View Figure 1
Further studies indicate that the process of STK33 expression is the same in mutant K-ras and wild cancer cell lines, and no STK33 mutation is detected by DNA sequence analysis; transfection of wild-type STK33 can't reverse the effect of K-ras genetic suppression on the survival and proliferation of these tumor cells. This suggests that STK33 remains the same in transcription and structure, and it functions like a normal gene [14]. However, a typical oncogene is activated by change of structure or certain overexpression, and promotes oncogenesis directly [15]. Consequently, the dependency of STK33 may be involved in the establishment of a new variant functional pathway, and reflect a genome specific vulnerability of K-ras mutant tumor cells, instead of the common features of Ras active cells.
Babij et al. refute the idea that inhibition of STK33 might be a useful therapeutic approach to target human mutant K-ras tumor [16]. In their studies, they found that down-regulation of K-ras decreases the survival of mutant K-ras cells. In contrast, the down-regulation or overexpression of STK33 has no effect on K-ras signaling or survival of these cells. Analogously, a synthetic lethal siRNA (small interfering RNA) screen operated in a broad panel of K-ras wild-type or mutant cells shows that K-ras is essential for survival instead of STK33. By using high-throughput screening, they also obtained similar negative results with small molecule inhibitors of the STK33 kinase. The study indicates that some STK33 inhibitors show no synthetic lethal with Ras-dependent tumor cells [17,18]. Tian et al. found that the STK33/c-Myc association is more important in HCC cell proliferation rather than the kinase activity of STK33 [19]. Currently, these conclusions remain controversial, which reflects the complexity of STK33 function [20].
Mechanism Underlying the Action of STK33 in Cancer Cells
So far, the mechanism of how STK33 influences the mutant K-ras tumor cells is still unclear. Studies have shown that ribosomal protein S6 kinase 1 (S6K1) is an important factor in signal transduction pathway. It can phosphorylate the pro-apoptotic BH3-only protein BAD at serine 136 site, which will result in its inactivation and suppresses the apoptosis in the path of mitochondrion [14]. Research indicates that apoptosis caused by STK33 suppression is mediated via the mitochondrial pathway; suppression of STK33 can reduce the phosphorylation of S6K1 of mutant K-ras tumor cells, thereby relieving the phosphorylation of Bad136 and inducing cell apoptosis. Thus, the mechanism by which STK33 suppression reduces cell apoptosis is related to mitochondrial apoptosis activated by inducing BAD [21,22]. In most tumor cells, PI3K (phosphatidylinositol 3-kinase) /AKT/mTOR (mammalian target of rapamycin) is activated by the mutation of its components or the activation of the upstream signal molecules, thus causing the dysregulation of cell proliferation, abnormity of anti-apoptosis and metabolism [23,24]. As a downstream effector of mammalian target of rapamycin complex 1 (mTORC1), S6K1 may act within the PI3K/AKT cascade or the mitogen-activated protein kinase (MAPK) signaling pathway [25], which increases mTORC1 activity.
It is reported that STK33 could bind to c-Myc directly and increase c-Myc transcription activity. In particular, the C-terminus of STK33 blocked STK33/c-Myc association, downregulated c-Myc transcription activity, and reduced HCC cell proliferation in vitro and in vivo [19].
Recently, we found [26] that STK33-RNA knockdown led to an increase in Caspase-3, Nm-23-H1 and E-Cadherin expressions and a reduction in Bcl-2, Ki-67 and Vimentin expressions. Moreover, PD98059 significantly reduced both ERK1/2 and STK33 expressions in Fadu cells. This indicates that STK33 is a potential oncogene and a promising diagnostic marker for HSCC. STK33 may promote tumorigenesis and progression of HSCC, and serve as a valuable molecular target for treatment of HSCC.
Conclusion
STK33 gene encodes a serine/threonine kinase, which is identified as a member of the CAMK family. Because it is involved in the “synthetic lethal” process of Ras depend tumor cells, it has attracted considerable interest in recent years. However, other research has refuted the conclusion that STK33 is a potential target for treating human mutant KRAS tumors. These conflicting reports demonstrate that some controversies regarding STK33 remain and indicate the complexity of STK33 function. Moreover, it may be employed as a promising diagnostic marker and a potential therapeutic target for the many kinds of tumors.
Acknowledgement
We would like to thank Dr. Luhua Zhang of Stanford University and Dr. Wayne Johnson for revising the manuscript.
References
-
Molli PR, Li DQ, Murray BW, Rayala SK, Kumar R (2009) PAK signaling in oncogenesis. Oncogene 28: 2545-2555.
-
Singh A, Settleman J (2009) Oncogenic K-ras "addiction" and synthetic lethality. Cell Cycle 8: 2676-2677.
-
Downward J (2009) Finding the weakness in cancer. N Engl J Med 361: 922-924.
-
Mujica AO, Hankeln T, Schmidt ER (2001) A novel serine/threonine kinase gene, STK33, on human chromosome 11p15.3. Gene 280: 175-181.
-
Ishida A, Sueyoshi N, Shigeri Y, Kameshita I (2008) Negative regulation of multifunctional Ca(2+)/calmodulin-dependent protein kinases: physiological and pharmacological significance of protein phosphatases. British Journal of Pharmacology 154: 729-740.
-
Wayman GA, Tokumitsu H, Davare MA, Soderling TR (2011) Analysis of CaM-kinase signaling in cells. Cell Calcium 50: 1-8.
-
Mujica AO, Brauksiepe B, Saaler-Reinhardt S, Reuss S, Schmidt ER (2005) Differential expression pattern of the novel serine/threonine kinase, STK33, in mice and men. FEBS J 272: 4884-4898.
-
Kamata A, Takeuchi Y, Fukunaga K (2006) Identification of the isoforms of Ca2+/calmodulin-dependent protein kinase II and expression of brain-derived neurotrophic factor mRNAs in the substantia nigra. Journal of Neurochemistry 96: 195-203.
-
Skelding KA, Rostas JA, Verrills NM (2011) Controlling the cell cycle: the role of calcium/calmodulin-stimulated protein kinases I and II. Cell Cycle 10: 631-639.
-
Brauksiepe B, Mujica AO, Herrmann H, Schmidt ER (2008) The Serine/threonine kinase Stk33 exhibits autophosphorylation and phosphorylates the intermediate filament protein Vimentin. BMC biochemistry 9: 25.
-
Brauksiepe B, Baumgarten L, Reuss S, Schmidt ER (2014) Co-localization of serine/threonine kinase 33 (Stk33) and vimentin in the hypothalamus. Cell Tissue Res 355: 189-199.
-
Le Meur N, Gentleman R (2008) Modeling synthetic lethality. Genome Biol 9: R135.
-
O'Brien T, Stokoe D (2009) Converting cancer mutations into therapeutic opportunities. EMBO Mol Med 1: 297-299.
-
Scholl C, Fröhling S, Dunn IF, Schinzel AC, Barbie DA, et al. (2009) Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell 137: 821-834.
-
Luo J, Solimini NL, Elledge SJ (2009) Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 136: 823-837.
-
Babij C, Zhang Y, Kurzeja RJ, Munzli A, Shehabeldin A, et al. (2011) STK33 kinase activity is nonessential in KRAS-dependent cancer cells. Cancer Res 71: 5818-5826.
-
Luo T, Masson K, Jaffe JD, Silkworth W, Ross NT, et al. (2012) STK33 kinase inhibitor BRD-8899 has no effect on KRAS-dependent cancer cell viability. Proc Natl Acad Sci USA 109: 2860-2865.
-
Weïwer M, Spoonamore J, Wei J, Guichard B, Ross NT, et al. (2012) A Potent and Selective Quinoxalinone-Based STK33 Inhibitor Does Not Show Synthetic Lethality in KRAS-Dependent Cells. ACS Med Chem Lett 3: 1034-1038.
-
Yang T, Song B, Zhang J, Yang GS, Zhang H, et al. (2016) STK33 promotes hepatocellular carcinoma through binding to c-Myc. Gut 65: 124-133.
-
Fröhling S, Scholl C (2011) STK33 kinase is not essential in KRAS-dependent cells--letter. Cancer Res 71: 7716.
-
Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, et al. (2009) A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 137: 835-848.
-
Singh A, Greninger P, Rhodes D, Koopman L, Violette S, et al. (2009) A gene expression signature associated with "K-Ras addiction" reveals regulators of EMT and tumor cell survival. Cancer Cell 15: 489-500.
-
Cao P, Maira SM, Garcia-Echeverria C, Hedley DW (2009) Activity of a novel, dual PI3-kinase/mTor inhibitor NVP-BEZ235 against primary human pancreatic cancers grown as orthotopic xenografts. British Journal of Cancer 100: 1267-1276.
-
Wu X, Kihara T, Akaike A, Niidome T, Sugimoto H (2010) PI3K/Akt/mTOR signaling regulates glutamate transporter 1 in astrocytes. Biochemical and biophysical research communications 393: 514-518.
-
Zhou H, Huang S (2010) The complexes of mammalian target of rapamycin. Curr Protein Pept Sci 11: 409-424.
-
Huang L, Chen C, et al. (2015) STK33 overexpression in hypopharyngeal squamous cell carcinoma: possible role in tumorigenesis. BMC Cancer 15: 13.
