

International Journal of Stem cell Research and Therapy
Advantage in Human Induced Pluripotent Stem Cells Research of X-Linked Genetic Diseases for Drug Screening
Takeo Kubota*
Department of Epigenetic Medicine, Graduate School of Interdisciplinary Research, University of Yamanashi, Japan
*Corresponding author: Takeo Kubota, Department of Epigenetic Medicine, Graduate School of Interdisciplinary Research, University of Yamanashi, 1110 Shimokato, Chuo, Yamanashi, 409-3898, Japan, Tel: +81-55-273-9557, Fax: +81-55-273-9561, E-mail: takeot@yamanashi.ac.jp
Int J Stem Cell Res Ther, IJSCRT-2-012, (Volume 2, Issue 2), Short Review; ISSN: 2469-570X
Received: September 02, 2015 | Accepted: October 16, 2015 | Published: October 22, 2015
Citation: Kubota T (2015) Advantage in Human Induced Pluripotent Stem Cells Research of X-Linked Genetic Diseases for Drug Screening. Int J Stem Cell Res Ther 2:012. 10.23937/2469-570X/1410012
Copyright: © 2015 Kubota T. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Human induced pluripotent stem cell (hiPSC) technology has widely been used for modeling of various genetic diseases. In the disease-modeling iPSC studies, it is necessary to generate hiPSC lines from peripheral tissues of multiple patients and of multiple age- and sex-matched control individuals in order to minimize the differences in genetic background that can affect the results. However, we can generate disease model-hiPSC line with mutant allele being expressing and normal control-hiPSC line with normal allele being expressing from a patient with an X-linked dominant disease such as Rett syndrome, taking advantage of use of the genetic nature (X-chromosome inactivation) and the hiPSC nature (monoclonal proliferation). Furthermore, it may be a straightforward way to identify drug candidates that normalize or minimize the abnormal features and gene expression patterns by a comparative study between mutant active- and normal active-hiPSC lines. Such drug-screening strategy can also be applicable to more major X-linked recessive disease such as Duchenne muscular dystrophy and Fabry disease by generating hiPSC lines from fibroblasts from the heterozygous females. Therefore, X-linked diseases may be efficient targets for drug screening using hiPSC derived from the patients, since faster drug screening hiPSC studies can be conducted for X-linked diseases compared with autosomal diseases by utilizing the genetic natures and the hiPSC nature.
Keywords
Human induced pluripotent stem cells; HiPSC; X-linked disease; X-chromosome inactivation; Genetic background; Drug screening
Short Review
Human induced pluripotent stem cell (hiPSC) technology has widely been used for modeling of various genetic diseases, especially neurological disorders, in which the target brain tissue is difficult to obtain from the patients [1,2]. In the disease-modeling hiPSC studies, it is necessary to generate iPSC lines from peripheral tissues of multiple patients and of multiple age- and sex-matched control individuals in order to minimize the differences in genetic background that can affect the results. Recent genome editing technology by ZFN, TALEN and CRISPR methods may overcome the issue of genetic background difference, because a certain patient-specific mutation can intentionally be introduced into the genome of a normal iPSC line and can clarify disease-specific nature caused by the mutation [3,4].
In an X-linked dominant disease, patients are all female since male patients are embryonic lethal. The female patients with an X-linked dominant disease have two types of cells: the cells with X chromosome harboring wild type (normal) gene being active and the cells with X chromosome harboring mutant gene being active (Figure 1). This is caused by random X chromosome inactivation (XCI), which is a genetic phenomenon that one of two X chromosome in females is randomly inactivated for dosage compensation of X-linked genes between males and females.
In the study, the two patients (monozygotic twins) with Rett syndrome (RTT) characterized by epilepsy, ataxic gait and autism shared the same mutation (one base deletion) in X-linked Methyl-CpG binding protein 2 (MECP2) gene. Random XCI patterns were demonstrated in their skin fibroblasts of two RTT patients by a DNA methylation based XCI assay [5]. We then generated iPSC from the skin fibroblast cell lines from the two patients using standard methods and transduction of OCT4-, SOX2-, KLF4- and c-MYC- containing retroviruses [6], and found extremely non-random XCI patterns in the majority of iPSC colonies due to monoclonal proliferation of iPSC cells as previously reported [7]. Based on such XCI phenomenon, we successfully obtained two types of iPSC line from the patients: the line with X chromosome harboring the maternally-derived normal MECP2 allele being active and the line with X chromosome harboring the paternally-derived mutant MECP2 allele being active (Figure 1). We then compared gene expression patterns between two types of the iPSC-derived neural cell lines, and found that the genes associated with astrocyte development, such as GFAP, were aberrantly expressed and thus the proportion of GFAP positive cells were increased in the mutant MECP2 active iPSC-derived neural cell line, compared with the normal MECP2 active line in both of the two patients [6]. These results, together with previous findings [8,9], indicate that not only abnormal function of astrocyte due to lack of MeCP2 but also abnormal proportion of astrocyte-like neural cells may contribute to the pathogenesis of RTT.
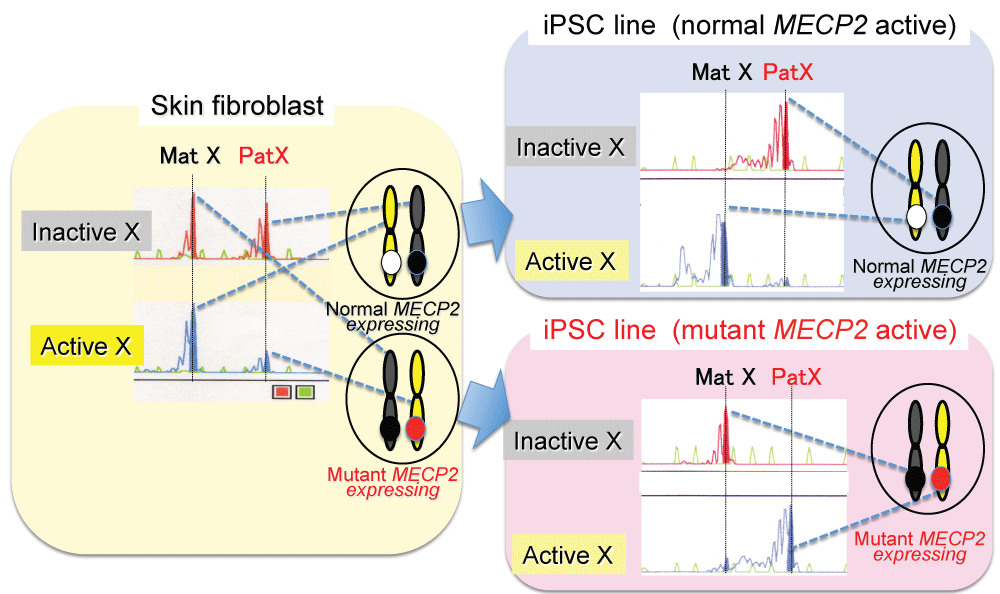
.
Figure 1: Two types of iPSC line generated from patients with Rett syndrome (an X-linked dominant genetic disease).
An X chromosome inactivation (XCI) assay showed a random XCI pattern in skin fibroblasts of the patient, indicating that the fibroblast contained the cells with X chromosome harboring normal MECP2 being active and the cells with X chromosome harboring mutant MECP2 being active (left). The assay showed two types of extremely non-random XCI patterns in iPSC lines: the line with X chromosome harboring normal MECP2 being active (normal MECP2 active) (right above), with X chromosome harboring mutant MECP2 being active (mutant MECP2 active) (right below).
View Figure 1
As mentioned above, we could generate the disease-model mutant active iPSC lines and the normal control iPSC lines with the same genetic background from patients with X-linked disease. This may be an advantage of X-linked diseases to autosomal diseases for identification of the disease-specific phenotypes (e.g., cell function, gene expression pattern) and for unnecessariness to analyze hiPSC lines from multiple patients' and normal individuals. Furthermore, it may be a straightforward way to identify drug candidates that normalize or minimize the abnormal features and gene expression patterns by a comparative study between mutant active- and normal active-hiPSC lines.
The applicants of the drug-screening hiPSC strategy are not only rare X-linked dominant diseases such as RTT but also major X-linked recessive disease such as Duchenne muscular dystrophy and Fabry disease by generating hiPSC lines from fibroblasts from the heterozygous females regardless of having clinical symptoms [10,11].
In conclusion, the authors would like to propose that X-linked dominant and recessive diseases are efficient targets for drug screening using hiPSC derived from the patients' peripheral tissues, since hiPSC-based drug screening studies can be performed faster in X-linked diseases, compared with autosomal diseases, by utilizing the genetic natures of XCI and the hiPSC nature of monoclonal proliferation.
Acknowledgements
The author thank Drs. Tomoko Ando-Noda, Wado Akamatsu, Kunio Miyake, Takuya Matsumoto, Ryo Yamaguchi, Tsukasa Sanosaka, Yohei Okada, Tetsuro Kobayashi, Manabu Ohyama, Kinichi Nakashima, Hiroshi Kurosawa and Hideyuki Okano for the original hiPSC research, and Japan Agency for Medical Research and Development (AMED) for funds for the development of core technologies for innovative drug development based upon IT, and the Ministry of Education, Science, Sports and Culture (MEXT), Grants-in-Aid (KAKENHI) for Scientific Research (B) (#26293245) and Exploratory Research (#15K15388).
References
-
Soldner F, Hockemeyer D, Beard C, Gao Q, Bell GW, et al. (2009) Parkinson's disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 136: 964-977.
-
Imaizumi Y, Okano H (2014) Modeling human neurological disorders with induced pluripotent stem cells. J Neurochem 129: 388-399.
-
Soldner F, Laganiere J, Cheng AW, Hockemeyer D, Gao Q, et al. (2011) Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell 146: 318-331.
-
Wu J, Darabi R (2014) Impact of new genome editing tools on iPS cell based therapies. Int J Stem Cell Res Ther 1: 1.
-
Kubota T, Nonoyama S, Tonoki H, Masuno M, Imaizumi K, et al. (1999) A new assay for the analysis of X-chromosome inactivation based on methylation-specific PCR. Hum Genet 104: 49-55.
-
Andoh-Noda T, Akamatsu W, Miyake K (2015) Differentiation of multipotent neural stem cells derived from Rett syndrome patients is biased toward the astrocytic lineage. Mol Brain 8: 31.
-
Tchieu J, Kuoy E, Chin MH, Trinh H, Patterson M, et al. (2010) Female human iPSCs retain an inactive X chromosome. Cell Stem Cell 7: 329-342.
-
Lioy DT, Garg SK, Monaghan CE, Raber J, Foust KD, et al. (2011) A role for glia in the progression of Rett's syndrome. Nature 475: 497-500.
-
Williams EC, Zhong X, Mohamed A, Li R, Liu Y, et al. (2014) Mutant astrocytes differentiated from Rett syndrome patients-specific iPSCs have adverse effects on wild-type neurons. Hum Mol Genet 23: 2968-2980.
-
Shoji E, Sakurai H, Nishino T, Nakahata T, Heike T, et al. (2015) Early pathogenesis of Duchenne muscular dystrophy modelled in patient-derived human induced pluripotent stem cells. Sci Rep 5: 12831.
-
Kawagoe S, Higuchi T, Otaka M, Shimada Y, Kobayashi H, et al. (2013) Morphological features of iPS cells generated from Fabry disease skin fibroblasts using Sendai virus vector (SeVdp). Mol Genet Metab 109: 386-389.
