

International Journal of Stem cell Research and Therapy
Regulation of Normal Somatic Cell and Cancer Cell Reprogramming by p53
Jie Yuan1,2 and Qin Yang1*
1Cancer Biology Division, Washington University School of Medicine, Saint Louis, MO 63108, USA
2School of Medicine, Jinan University, Guangzhou 510632, China
*Corresponding author: Qin Yang, MD, PhD, Cancer Biology Division, Washington University School of Medicine, 4511 Forest Park, St. Louis, MO 63108, USA, Tel: 314-747-5445; Fax: 314-362-9790; E-mail: qyang@wustl.edu
Int J Stem Cell Res Ther, IJSCRT-2-016, (Volume 2, Issue 2), Review Article; ISSN: 2469-570X
Received: November 25, 2015 | Accepted: December 19, 2015 | Published: December 24, 2015
Citation: Yuan J, Yang Q (2015) Regulation of Normal Somatic Cell and Cancer Cell Reprogramming by p53. Int J Stem Cell Res Ther 2:016. 10.23937/2469-570X/1410016
Copyright: © 2015 Yuan J, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Reprogramming healthy somatic cells into disease-relevant cell types through cellular reprogramming has been intensively investigated. The discovery of reprogramming methods holds the promise of generating desired cells for disease modeling, drug screening studies and treatment of numerous diseases. Recently studies also focus on different disease cell reprogramming, including cancer cell reprogramming. Reprogramming and tumorigenesis share many similarities and the tumor suppressor p53 suppresses both processes. Understanding of the roles of p53 in somatic cell and cancer cell reprogramming could illuminate molecular mechanisms underlying the pathogenesis of human cancer and develop novel strategies for cell replacement therapy and cancer treatment.
Keywords
p53, Reprogramming, Cancer, iPS
Introduction
The ability to convert somatic cells into disease-relevant cell types through cellular reprogramming has opened new doors for basic research and regeneration medicine1. In the past, there were no reliable methods for converting somatic cells into another type of cells. Takahashi et al. demonstrated that defined factors could drive skin-derived fibroblasts to induced pluripotent stem (iPS) cells that could be further differentiated into the desired cell type [1].
Reprogramming healthy somatic cells into iPS cells with defined factors has been intensively investigated. However, reprogramming cancer cells has comparatively been lagging behind [2-4]. Direct reprogramming cancer cells into normal cells is an innovative strategy for cancer treatment. Recent reports have showed that defined transcription factors can induce reprogramming of cancer cells into iPS cells, supporting this notion [2-4]. The tumor suppressor p53 is considered today the most important tumor suppressor gene in humans [5,6] and its inactivation or mutations are the most common in cancers. Reprogramming and tumorigenesis are stepwise processes that share many similarities and p53 suppresses both reprogramming and tumorigenesis [2-4,7,8]. In this review, we discuss the roles of p53 in normal somatic cell and cancer cell reprogramming (Figure 1).
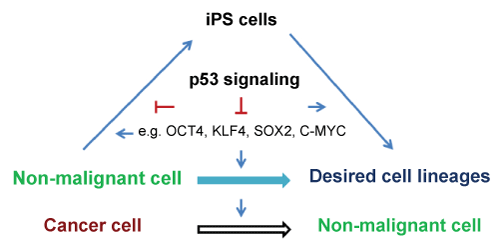
.
Figure 1: Schematic drawing of a proposed model for effects of p53 on somatic cell and cancer cell reprogramming.
View Figure 1
p53 Regulates Pluripotency Reprogramming
Differentiated somatic cells have been reprogrammed to a pluripotent state by forced expression of a set of transcription factors [1], indicating that terminally differentiated cells can be induced to undergo cell fate change. p53 reduces cancer initiation by regulating apoptosis, DNA repair, cell cycle and senescence, contributing to its main role as the "guardian of the genome". Recent studies show new roles of p53 in a wide range of processes, including cell self-renewal, differentiation, and cell fate decisions [9-11]. Several lines of evidence suggested that p53 governs the quantity and quality of various stem cells through regulation of self renewal ability and differentiation. One example is that the p53 protein can be phosphorylated and suppress the transcription of Nanog, leading to stem cells to lose self renewal potential and then differentiation [12]. Many groups reported that p53 has been shown to inhibit reprogramming of fibroblasts to iPS cells [7,8,13-17]. Yamanaka et al. found that c-Myc induces p53-dependent apoptosis in fibroblasts, leading to a reduced rate of reprogramming [16]. Zhao et al. showed that depletion of p53 combined with overexpression of Utf1 dramatically increased in iPS cell formation [17]. Other groups also provided strong evidence that p53 serves as a potent barrier to somatic cell reprogramming and dramatically reduces the efficiency of dedifferentiation [12-14].
Mechanistically, the overexpression of exogenous transcription factors is thought to activate p53, which might in turn lead to cell cycle arrest and apoptosis. In addition, during pluripotency induction, cells presenting with DNA damage and chromosomal abnormalities will be excluded from becoming iPS cells. A report showed that p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity [18]. Another report showed that in the absence of p53, cells with a defective DNA repair pathway could undergo reprogramming, allowing the generation of iPS cells with genomic instability [19]. Hanna et al. showed that an additional role of p53 in reprogramming may be an indirect effect on cell proliferation [20]. Sarig et al. examined the role of p53 mutant in pluripotency induction and found that mutant p53 has a gain-of-function in reprogramming. p53 mutant mouse embryonic fibroblasts (MEFs) were reprogrammed more efficiently than MEFs derived from both wild-type and p53 knockout mice, indicating that mutant p53 actually increases reprogramming efficiency beyond that facilitated by the absence of p53 alone. Thus, these reports suggest that suppressing p53 regulates apoptosis, DNA repair, senescence, and cell cycle, thereby increasing reprogramming efficiency.
p53 Regulates Direct Lineage Reprogramming
The field of direct somatic lineage conversion bypass iPS cells has already attracted much attention. Wernig's group demonstrated that a set of neural factors can directly convert fibroblasts into neurons [21,22]. Several laboratories have used various neural factors and microRNAs to generate fibroblast-neuron conversion [23-30]. Furthermore, recent studies have reduced the numbers of transcription factors required for neuronal trans-differentiation by including small molecules to promote neuronal phenotypes [31,32]. Direct fibroblast-neuron conversion provides an alternative, potentially complementary, tool to many of the proposed applications of iPS technology for both disease modeling and development of cell-based therapies. This approach has a number of advantages, including: the time required to generate, expand and differentiate pluripotent cells is avoided, and the postmitotic state of induced cells has a much lower risk of cancer and teratoma formation.
Delineating the molecular mechanism behind somatic cell reprogramming will greatly aid further development of the method. In our recent studies, we have demonstrated that suppression of p53 or cellular senescence is a key step in the direct human somatic cell reprogramming [33,34]. We found that inhibition of p53 efficiently induces conversion of fibroblasts to neurons without introduction of any transcription factors. Knockout or knock-down of p53 efficiently reprogrammed fibroblasts into tri-neural cells by regulation of a set of neural transcription factors. The induction was specific to p53 depletion, because overexpression of wild-type p53 in cells expressing p53 shRNA essentially abolished fibroblast-neuron conversion. Furthermore, we found that mutant p53 lost the inhibitory function on reprogramming. Overexpression of a p53 mutant, R273H, in which Arg 273 in the DNA binding domain was replaced with His [35-37], did not affect the induction time course, indicating that this p53 mutant has lost the inhibitory function in reprogramming. In addition, we provided a new method to convert most fibroblasts to neurons within only a week of time. Moreover, the defined transcription factors failed in inducing fibroblast-neuron conversion in late passage fibroblasts, while depleting p53 is advantageous in inducing neuron in both early and late passage fibroblast cells.
p53 Regulates Cancer Cell Reprogramming
One critical question in cancer research is: are cancer cells capable of reprogramming from their malignant state? The iPS reprogramming strategy might provide an opportunity that human cancer cells can be similarly reprogrammed and subsequently differentiated with loss of tumorigenicity. Although unidentified biological barriers may exist [38-40], reprogramming of both solid and liquid tumors to iPS cells has been reported by different groups [39,41-50]. Loss of tumorigenicity by unknown mechanisms and induced de-differentiation to pluriopotency seem to be common features of reprogrammed cells from different cancers [51-55]. Zhang et al. found that reprogramming with defined factors induces sarcoma cells to lose tumorigenicity, come back to a "normal state" and undergo differentiation into different terminal "normal" cells [41]. The epigenetic regulation of oncogene c-Myc might have a key role in the process of reprogramming sarcoma cells. These studies may, therefore, provide a novel strategy for cancer treatment via pluripotency induction.
Because most cancers have shown the suppression of the p53 signaling pathways, one would expect accelerated cancer cell reprogramming with p53 inactivation. Studies on the link between p53 and cancer cell differentiation were first published about 20 years ago [56-59]. Those reports showed that the de-differentiated phenotype of cancers correlates with p53 loss and increased tumorigenesis. Recently, Telerman et al. found that p53 plays a key role in cancer cell reprogramming through interaction with TPT1/TCTP (encoding translationally controlled tumor protein) [60,61]. They have previously established cancer reversion models and identified ~300 genes putatively implicated in reversion, including TPT1/TCTP [62]. TPT1/TCTP regulates the p53-MDM2-Numb axis in cancer cell programming. Another study showed that gain-of-function of mutant p53 enhances reprogramming efficiency and the Myc pathway cooperates with a p53 mutant protein to disrupt the efficiency of reprogramming [63]. Using a temperature sensitive mutant of the p53 gene, Levine and colleagues examined the impact of the temporal expression of wild type p53 in preventing stem cell induction from somatic and cancer cells [63]. Reactivation of p53 during the reprogramming process not only interrupted the formation of iPS cells, but also induced newly formed stem cells to differentiate. Moreover, p53 mutants showed differential effects on the stem cell reprogramming efficiency in a c-Myc dependent manner. Although both responded to the inhibition of reprogramming by the p53 protein, somatic cells and cancer cells are different from each other in several ways.
Possible Mechanisms of p53 in Reprogramming
Although current studies suggest that p53 regulates apoptosis, DNA repair, senescence, and cell cycle in reprogramming, the mechanisms by which the p53 protein inhibits normal and cancer cell reprogramming are largely unknown. To detect the p53 signaling pathway, Levine and colleagues found that p21 (Cdkn1a), but not Puma (Bbc3) played a partial role in iPS cells formation probably by slowing cell division [63]. They also found that activation of p53 functions in iPS cells induced senescence and differentiation in stem cell populations. To examine epigenetic alterations of DNA, they found increases in DNA methylation at the IGF2-H19 loci with high rate of birth defects in female offspring of p53 knockout mice, indicating that p53 knockout mice would display epigenetic alterations during embryonic development and thus resulted in birth defects. These results suggested that a portion of p53 ability to inhibit reprogramming may come from its enforcement of preventing epigenetic changes.
p21 is one of the most prominent targets of p53 involved in regulation of cell-cycle [64,65] and iPS reprogramming [9,13,15]. To determine whether p53 depletion induced fibroblast-neuron conversion by inhibiting the p53-p21 signaling pathway, we inhibited p21 and did not observe neuronal morphology in shp21-expressing cells [34]. Furthermore, co-expression of shp21 or wild-type p21 with shp53 did not affect shp53-induced fibroblast-neuron conversion in IMR90 cells. These data indicate that p21 is not involved in shp53-induced fibroblast-neuron conversion.
Global gene expression analysis reveals that the gene profiling in induced cells is significantly changed, compared with that in the parental fibroblasts [33,34]. Moreover, p53 plays a key role through regulating a set of transcription factors during the reprogramming process, supporting the notion that p53 may act as a "master switch" in reprogramming. We found that p53 bound to the promoter DNA of neurogenic transcription factor Neurod2 and regulated its expression during fibroblast-neuron conversion [34]. Telerman et al. found that TPT1/TCTP and p53 are involved in a reciprocal negative-feedback loop in cancer cell reprogramming [60,61]. TPT1/TCTP inhibits MDM2 auto-ubiquitination and promotes p53 degradation by competing Numb for binding to p53-MDM2-containing complexes. In addition, p53 directly represses TPT1/TCTP transcription. Since TPT1/TCTP is a highly significant prognostic factor in breast cancer, targeting TPT1/TCTP in cancer cell reprogramming could open a new route to cancer treatment.
Lineage conversions between very distantly related cell types might involve two main steps: 1) reprogramming of prior donor cells into induced progenitors, which might not pass through a pluripotent state; 2) subsequent re-differentiation into a complete and functional lineage. Depletion of p53 enhanced reprogramming suggests that p53 may inhibit both steps (Figure 1). Consistent with this view, inhibition of p53 enhances both iPS cell reprogramming and neural conversion [33,34], implicating a general mechanism of reprogramming where inhibiting p53 may generate and develop complete and functional lineages. One hypothesis is that depletion of p53 prevents loss of proliferative potential in cell cycle-arrested cells and that it can therefore transform irreversible arrest into a reversible condition. When the cell-cycle is arrested by neural induction medium (for fibroblast-neuron conversion) or reprogramming factors (for iPS cell formation), in the absence of p53, the cells remain quiescent but not senescent because they retain the capacity to resume proliferation. Under defined conditions, the cells are reprogrammed into progenitors and sequentially re-differentiated into neural lineages or continue to reprogram into iPS cells.
Concluding remarks
Somatic cell conversion by introduction of defined factors might have significant implications for understanding critical processes for development, in vitro disease modeling and cell replacement therapies, although generally with low percentages and very slow time course of reprogramming [66-68]. Understanding the role of p53 in reprogramming may have wide-spread impact on our understanding and development of cell replacement therapy. Although the link between p53 and tumorigenicity makes using the induced cells lacking p53 impractical, treating cells during reprogramming with reversible compounds to promote immortalization transiently is feasible. In the future, testing compounds that block p53 downstream factors for cell reprogramming may be a novel approach for maximizing the generation of "safe" cells for clinical applications, including for old people who may suffer more disorders. We believe that the p53 pathway will be a key process to target in pursuit of finding more efficient strategies for generating desired cells.
Reprogramming and tumorigenesis are connected processes that share many similarities. Targeting the p53 pathways underlying the direct reprogramming of cancer cells opens new perspectives for understanding of mechanisms of cancer pathogenesis and development of an alternative approach in cancer treatment.
Conflict of Interest
The authors declare that they have no conflict of interest.
Acknowledgment
This work is supported in part by grants from the Natural Science Foundation (No. 81228017) and Innovation Award at Washington University.
References
-
Takahashi K et al. (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 1: 861-872.
-
Klimczak M (2015) Oncogenesis and induced pluripotency - commonalities of signalling pathways. Contemp Oncol (Pozn) 19: A16-21.
-
Lin YC, Murayama Y, Hashimoto K, Nakamura Y, Lin CS, et al. (2014) Role of tumor suppressor genes in the cancer-associated reprogramming of human induced pluripotent stem cells. Stem Cell Res Ther 5: 58.
-
Wasik AM, Grabarek J, Pantovic A, Cieslar-Pobuda A, Asgari HR, et al. (2014) Reprogramming and carcinogenesis--parallels and distinctions. Int Rev Cell Mol Biol 308: 167-203.
-
Hofseth LJ, Robles AI, Yang Q, Wang XW, Hussain SP, et al. (2004) p53: at the crossroads of molecular carcinogenesis and molecular epidemiology. Chest 125: 83S-5S.
-
Hollstein M, Hergenhahn M, Yang Q, Bartsch H, Wang ZQ, et al. (1999) New approaches to understanding p53 gene tumor mutation spectra. Mutat Res 431: 199-209.
-
Menendez S, Camus S, Izpisua Belmonte JC (2010) p53: guardian of reprogramming. Cell Cycle 9: 3887-3891.
-
Tapia N, Schöler HR (2010) p53 connects tumorigenesis and reprogramming to pluripotency. J Exp Med 207: 2045-2048.
-
Lanni C, Racchi M, Memo M, Govoni S, Uberti D (2012) p53 at the crossroads between cancer and neurodegeneration. Free Radic Biol Med 52: 1727-1733.
-
Hede SM, Nazarenko I, Nistér M, Lindström MS (2011) Novel Perspectives on p53 Function in Neural Stem Cells and Brain Tumors. J Oncol 2011: 852970.
-
Mendrysa SM, Ghassemifar S, Malek R (2011) p53 in the CNS: Perspectives on Development, Stem Cells, and Cancer. Genes Cancer 2: 431-442.
-
Lin T, Chao C, Saito S, Mazur SJ, Murphy ME, et al. (2005) p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol 7: 165-171.
-
Hong H, Takahashi K, Ichisaka T, Aoi T, Kanagawa O, et al. (2009) Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature 460: 1132-1135.
-
Utikal J, Polo JM, Stadtfeld M, Maherali N, Kulalert W, et al. (2009) Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature 460: 1145-1148.
-
Kawamura T, Suzuki J, Wang YV, Menendez S, Morera LB, et al. (2009) Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 460: 1140-1144.
-
Yamanaka S (2007) Strategies and new developments in the generation of patient-specific pluripotent stem cells. Cell Stem Cell 1: 39-49.
-
Zhao Y, Yin X, Qin H, Zhu F, Liu H, et al. (2008) Two supporting factors greatly improve the efficiency of human iPSC generation. Cell Stem Cell 3: 475-479.
-
Marión RM, Strati K, Li H, Murga M, Blanco R, et al. (2009) A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature 460: 1149-1153.
-
González F, Georgieva D, Vanoli F, Shi ZD, Stadtfeld M, et al. (2013) Homologous recombination DNA repair genes play a critical role in reprogramming to a pluripotent state. Cell Rep 3: 651-660.
-
Hanna J, Saha K, Pando B, van Zon J, Lengner CJ, et al. (2009) Direct cell reprogramming is a stochastic process amenable to acceleration. Nature 462: 595-601.
-
Pang ZP, Yang N, Vierbuchen T, Ostermeier A, Fuentes DR, et al. (2011) Induction of human neuronal cells by defined transcription factors. Nature 476: 220-223.
-
Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Südhof TC, et al. (2010) Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463: 1035-1041.
-
Qiang L, Fujita R, Yamashita T, Angulo S, Rhinn H, et al. (2011) Directed conversion of Alzheimer's disease patient skin fibroblasts into functional neurons. Cell 146: 359-371.
-
Caiazzo M, Dell'Anno MT, Dvoretskova E, Lazarevic D, Taverna S, et al. (2011) Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature 476: 224-227.
-
Pfisterer U, Kirkeby A, Torper O, Wood J, Nelander J, et al. (2011) Direct conversion of human fibroblasts to dopaminergic neurons. Proc Natl Acad Sci U S A 108: 10343-10348.
-
Yoo AS, Sun AX, Li L, Shcheglovitov A, Portmann T, et al. (2011) MicroRNA-mediated conversion of human fibroblasts to neurons. Nature 476: 228-231.
-
Ambasudhan R, Talantova M, Coleman R, Yuan X, Zhu S, et al. (2011) Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell 9: 113-118.
-
Son EY, Ichida JK, Wainger BJ, Toma JS, Rafuse VF, et al. (2011) Conversion of mouse and human fibroblasts into functional spinal motor neurons. Cell Stem Cell 9: 205-218.
-
Ring KL, Tong LM, Balestra ME, Javier R, Andrews-Zwilling Y, et al. (2012) Direct reprogramming of mouse and human fibroblasts into multipotent neural stem cells with a single factor. Cell Stem Cell 11: 100-109.
-
Kim J, Su SC, Wang H, Cheng AW, Cassady JP, et al. (2011) Functional integration of dopaminergic neurons directly converted from mouse fibroblasts. Cell Stem Cell 9: 413-419.
-
Ladewig J, Mertens J, Kesavan J, Doerr J, Poppe D, et al. (2012) Small molecules enable highly efficient neuronal conversion of human fibroblasts. Nat Methods 9: 575-578.
-
Liu ML, Zang T, Zou Y, Chang JC, Gibson JR, et al. (2013) Small molecules enable neurogenin 2 to efficiently convert human fibroblasts into cholinergic neurons. Nat Commun 4: 2183.
-
Sun CK, Zhou D, Zhang Z, He L, Zhang F, et al. (2014) Senescence impairs direct conversion of human somatic cells to neurons. Nat Commun 5: 4112.
-
Zhou D, Zhang Z, He LM, Du J, Zhang F, et al. (2014) Conversion of fibroblasts to neural cells by p53 depletion. Cell Rep 9: 2034-2042.
-
Goh AM, Coffill CR, Lane DP (2011) The role of mutant p53 in human cancer. J Pathol 223: 116-126.
-
Yang Q, Zhang R, Wang XW, Linke SP, Sengupta S, et al. (2004) The mismatch DNA repair heterodimer, hMSH2/6, regulates BLM helicase. Oncogene 23: 3749-3756.
-
Yang Q, Zhang R, Wang XW, Spillare EA, Linke SP, et al. (2002) The processing of Holliday junctions by BLM and WRN helicases is regulated by p53. J Biol Chem 277: 31980-31987.
-
Lang JY, Shi Y, Chin YE (2013) Reprogramming cancer cells: back to the future. Oncogene 32: 2247-2248.
-
Hochedlinger K, Blelloch R, Brennan C, Yamada Y, Kim M, et al. (2004) Reprogramming of a melanoma genome by nuclear transplantation. Genes Dev 18: 1875-1885.
-
Ramos-Mejia V, Fraga MF, Menendez P (2012) iPSCs from cancer cells: challenges and opportunities. Trends Mol Med 18: 245-247.
-
Zhang X, Cruz FD, Terry M, Remotti F, Matushansky I (2013) Terminal differentiation and loss of tumorigenicity of human cancers via pluripotency-based reprogramming. Oncogene 32: 2249-2260, 2260.
-
Carette JE, Pruszak J, Varadarajan M, Blomen VA, Gokhale S, et al. (2010) Generation of iPSCs from cultured human malignant cells. Blood 115: 4039-4042.
-
Utikal J, Maherali N, Kulalert W, Hochedlinger K (2009) Sox2 is dispensable for the reprogramming of melanocytes and melanoma cells into induced pluripotent stem cells. J Cell Sci 122: 3502-3510.
-
Miyoshi N, Ishii H, Nagai K, Hoshino H, Mimori K, et al. (2010) Defined factors induce reprogramming of gastrointestinal cancer cells. Proc Natl Acad Sci U S A 107: 40-45.
-
Chang G et al. (2010) Linking incomplete reprogramming to the improved pluripotency of murine embryonal carcinoma cell-derived pluripotent stem cells. PLoS One e10320.
-
Choi SM, Liu H, Chaudhari P, Kim Y, Cheng L, et al. (2011) Reprogramming of EBV-immortalized B-lymphocyte cell lines into induced pluripotent stem cells. Blood 118: 1801-1805.
-
Hu K, Yu J, Suknuntha K, Tian S, Montgomery K, et al. (2011) Efficient generation of transgene-free induced pluripotent stem cells from normal and neoplastic bone marrow and cord blood mononuclear cells. Blood 117: e109-119.
-
Lin SL, Chang DC, Chang-Lin S, Lin CH, Wu DT, et al. (2008) Mir-302 reprograms human skin cancer cells into a pluripotent ES-cell-like state. RNA 14: 2115-2124.
-
Amson R, Pece S, Lespagnol A, Vyas R, Mazzarol G, et al. (2011) Reciprocal repression between P53 and TCTP. Nat Med 18: 91-99.
-
Amson R, Karp JE, Telerman A (2013) Lessons from tumor reversion for cancer treatment. Curr Opin Oncol 25: 59-65.
-
Papp B, Plath K (2011) Reprogramming to pluripotency: stepwise resetting of the epigenetic landscape. Cell Res 21: 486-501.
-
Ho R, Chronis C, Plath K (2011) Mechanistic insights into reprogramming to induced pluripotency. J Cell Physiol 226: 868-878.
-
Han SS, Williams LA, Eggan KC (2011) Constructing and deconstructing stem cell models of neurological disease. Neuron 70: 626-644.
-
Cherry AB, Daley GQ (2012) Reprogramming cellular identity for regenerative medicine. Cell 148: 1110-1122.
-
Robinton DA, Daley GQ (2012) The promise of induced pluripotent stem cells in research and therapy. Nature 481: 295-305.
-
Yamaguchi T, Toguchida J, Wadayama B, Kanoe H, Nakayama T, et al. (1996) Loss of heterozygosity and tumor suppressor gene mutations in chondrosarcomas. Anticancer Res 16: 2009-2015.
-
Kemp CJ, Donehower LA, Bradley A, Balmain A (1993) Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell 74: 813-822.
-
Fagin JA, Matsuo K, Karmakar A, Chen DL, Tang SH, et al. (1993) High prevalence of mutations of the p53 gene in poorly differentiated human thyroid carcinomas. J Clin Invest 91: 179-184.
-
Donghi R, Longoni A, Pilotti S, Michieli P, Della Porta G, et al. (1993) Gene p53 mutations are restricted to poorly differentiated and undifferentiated carcinomas of the thyroid gland. J Clin Invest 91: 1753-1760.
-
Amson R, Pece S, Marine JC, Di Fiore PP, Telerman A (2013) TPT1/ TCTP-regulated pathways in phenotypic reprogramming. Trends Cell Biol 23: 37-46.
-
Amson R, Pece S, Lespagnol A, Vyas R, Mazzarol G, et al. (2011) Reciprocal repression between P53 and TCTP. Nat Med 18: 91-99.
-
Tuynder M, Susini L, Prieur S, Besse S, Fiucci G, et al. (2002) Biological models and genes of tumor reversion: cellular reprogramming through tpt1/TCTP and SIAH-1. Proc Natl Acad Sci U S A 99: 14976-14981.
-
Yi L, Lu C, Hu W, Sun Y, Levine AJ (2012) Multiple roles of p53-related pathways in somatic cell reprogramming and stem cell differentiation. Cancer Res 72: 5635-5645.
-
Abbas T, Dutta A (2009) p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer 9: 400-414.
-
Laiho M, Latonen L (2003) Cell cycle control, DNA damage checkpoints and cancer. Ann Med 35: 391-397.
-
Yang N, Ng YH, Pang ZP, Südhof TC, Wernig M (2011) Induced neuronal cells: how to make and define a neuron. Cell Stem Cell 9: 517-525.
-
Vierbuchen T, Wernig M (2012) Molecular roadblocks for cellular reprogramming. Mol Cell 47: 827-838.
-
Qiang L, Fujita R, Abeliovich A (2013) Remodeling neurodegeneration: somatic cell reprogramming-based models of adult neurological disorders. Neuron 78: 957-969.
