

International Journal of Stem cell Research and Therapy
Hypophosphatasia and Mesenchymal Stem Cells: A Therapeutic Promise
Maria Teresa Valenti1*, Luca Dalle Carbonare1 and Monica Mottes2
1Department of Medicine, Section of Internal Medicine, University of Verona, Italy
2Department of Neurological, Biomedical and Movement Sciences, Section of Biology and Genetics, University of Verona, Italy
*Corresponding author: Maria Teresa Valenti, PhD, Department of Medicine, Clinic of Internal Medicine, section D, University of Verona, Piazzale Scuro, 10, 37134 Verona, Italy, Tel: +39-045-8128450; Fax +39-045-8027496; E-mail: mariateresa.valenti@univr.it
Int J Stem Cell Res Ther, IJSCRT-3-020, (Volume 3, Issue 1), Review Article; ISSN: 2469-570X
Received: December 09, 2015 | Accepted: January 25, 2016 | Published: January 29, 2016
Citation: Valenti MT, Carbonare LD, Mottes M (2016) Hypophosphatasia and Mesenchymal Stem Cells: A Therapeutic Promise. Int J Stem Cell Res Ther 3:020. 10.23937/2469-570X/1410020
Copyright: © 2016 Valenti MT, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Hypophosphatasia (HPP) is due to mutations in ALPL gene which encodes the tissue non-specific alkaline phosphatase isozyme (TNSALP). Defective/inactive TNSALP causes an increased concentration of inorganic pyrophosphate (PPi) in bone matrix that impairs bone mineralization. The accumulation of extracellular PPi observed in HPP causes impairment in bone mineralization process and leads to a disturbance of calcium and Pi homeostasis. The pathogenesis of bone hypomineralization in HPP is relatively well understood; biomedical research aiming to treatment has been focused on the most obvious approach, i.e. enzyme replacement therapy, with unsatisfactory results. More innovative therapeutic approaches can be devised nowadays, thanks to current biotechnological innovations. Perspectively the use of mesenchymal stem cells (MSCs) represents an attractive approach for the treatment of HPP. MSCs, a population of adult stem cells, can differentiate into cells deriving from mesodermal lineage. Research in the field is progressively demonstrating their therapeutic capabilities in several skeleton-related disorders: we review a few recent applications in HPP patients.
Keywords
Hypophosphatasia, Mesenchymal stem cells
Background
HPP was first described in 1948 and has therefore been known for a long time as an inherited disorder of bone and mineral metabolism. HPP is a rare inborn error of metabolism due to mutations in ALPL gene located on chromosome 1 which encodes the tissue non-specific alkaline phosphatase isozyme. This gene is subject to high allelic heterogeneity and more than 260 ALPL missense mutations causing suboptimal activity of TNSALP have been identified. They are reported in a dedicated database (http://www.sesep.uvsq.fr/03_hypo_mutations.php). The first identified mutation was a homozygous missense in exon 6 of the ALPL gene, A162T.9. Later, new mutations, (i.e., R54C, R54P, E174K, Q190P, Y246H, D277A, D361V, Y419H, G317D2; E281K, A160T, F310L, G439R12, two frameshift mutations at positions 328 and 50313, respectively) were reported. Mornet reported 16 new missense mutations in European patients (i.e., S-1F, A23V, R58S, G103R, G112R, N153D, R167W, R206W, W253X, E274K, S428P, R433C, G456S, G474R and splice mutations in intron 6 and 9) [1,2].
ALPL mutations cause mineralization disorders including soft bones (rickets or osteomalacia) and defects in teeth as consequence of a reduced TNSALP activity.
The disease is classified by patient age when the first signs and symptoms manifest and by mode of inheritance (autosomal dominant/recessive): benign prenatal, lethal perinatal, infantile, childhood, adult and odonto- HPP [2,3]. The more severe forms of HPP (perinatal lethal/infantile) are transmitted as autosomal recessive traits and are due to ALPL mutations which impair TNSALP almost completely. Infantile HPP has been defined as a disease presenting symptoms before 6 months of age and Pyridoxine-responsive seizures often occur. The patients with the infantile form are affected also by respiratory complications and premature craniosynostosis due to the high intracranial pressure.
Several therapeutic approaches have been tested for patients with HPP including bone marrow, bone fragments and osteoblasts transplantation, parathyroid hormone administration, enzyme replacement therapy (ERT) with alkaline phosphatase (ALP)-rich serum obtained from patients affected by Paget's disease, infusion of plasma from healthy individuals or purified ALP [4,5]. The ERT, however, requires repeated subcutaneous administration of the enzyme, due to its short half-life in serum. Recently, clinical phase 2 studies with enzyme-replacement therapy with recombinant TNSALP have been perfomed [6]. Unfortunately, the variety of attempted treatments has obtained only limited clinical and radiographic improvements and no established therapeutic approach exists for HPP.
Bone formation
Endochondral bone formation is performed by osteoblasts that mineralize the extracellular matrix. This process includes the formation of crystalline hydroxyapatite in the matrix vesicles and matrix modulation to induce the propagation of apatite outside these vesicles. Among other factors, such as Ca2+ concentration and fibrillar collagen production, bone mineralization depends on the balance of inorganic phosphate (Pi) and inorganic pyrophosphate (PPi), a mineralization inhibitor. The osteoblastic molecules affecting bone mineral apposition by regulating the extracellular levels of PPi are TNSALP, NPP1 (nucleotide pyrophoshatase/phosphodiesterase isozyme) and the ANK gene product. In normal individuals, TNSALP functions as an ecto-enzyme, located on the outer surface of matrix vesicles of osteoblasts and chondrocytes where it hydrolizes the inorganic pyrophosphate (PPi), pumped extracellularly by the protein ANK or produced extracellularly by nucleoside triphosphate pyrophosphatase (NPP1) [2]. NPP1, a glycoprotein encoded by ENPP1 gene, is highly expressed by osteoblast and chondrocyte cells; it inhibits matrix mineralization by hydrolyzing extracellular nucleotides into inorganic pyrophosphate (PPi), a natural substrate of TNSALP [7] (Figure 1).
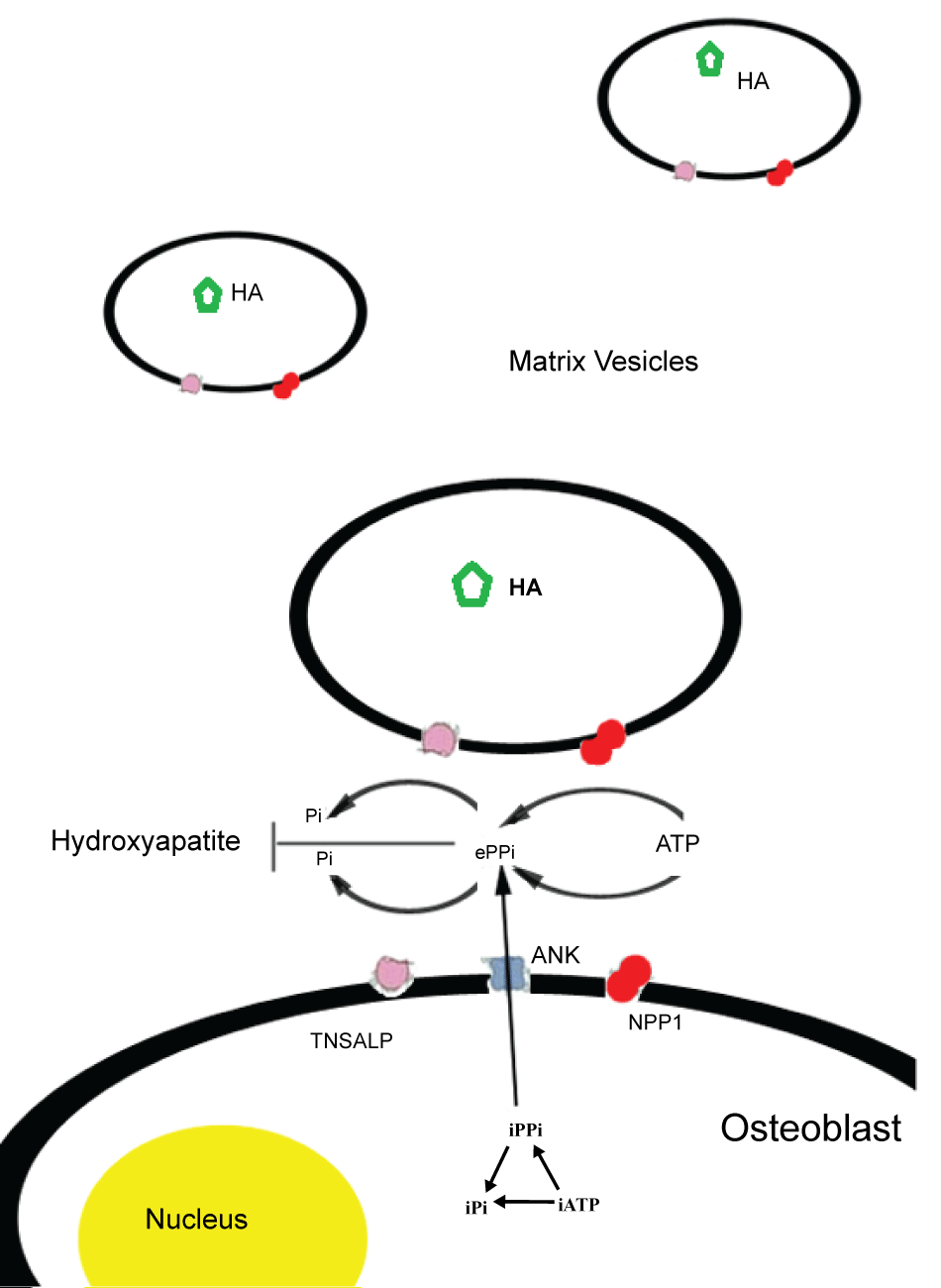
.
Figure 1: Schematic representation of molecules involved in Pi/PPi balance and mineralization process in matrix vesicles. TNSALP: Tissue non specific alkaline phosphatase, NPPI: Ectonucleotide Pyrophosphatase Phosphodiesterase 1, ANK: Ankylosis Protein, HA: Hydroxyapatite, Pi: Inorganic phosphate, PPi: Inorganic pyrophosphate.
View Figure 1
Many studies have shown that most of the ALPL mutations present in severe hypophosphatasia lead to the production of a mutant TNSALP protein unable to reach the cellular membrane, which accumulates in the Golgi apparatus and is subsequently degraded by the proteasome. In mild hypophosphatasia, on the contrary, the protein is to some extent- correctly located at the cell membrane.
The accumulation of extracellular PPi due to the reduced catalytic activity of mutant TNSALP found in HPP causes the pathogenetic block of bone mineralization and leads a disturbance of calcium and Pi homeostasis.
MSCs and bone diseases
Lately an increasing interest has arisen in the possible employment of MSCs for the treatment of bone defects. MSC contribution to bone fracture repair has been extensively documented; scaffolds seeded with MSCs are most often used in tissue engineering [8,9]. Treatments of bone diseases with MSCs have been tested in osteogenesis imperfecta, osteonecrosis of the femoral head, osteoporosis, rheumatoid arthritis, osteoarthritis and, recently, in HPP [10]. MSCs are multipotent cells and can differentiate in adipocytes, osteoblasts and chondrocytes. These cells can be selected from different tissues as bone marrow, dental pulp, adipose tissue, as well as placenta, amniotic fluid and umbilical cord blood [11]; they can be isolated from peripheral blood too [12]. When MSCs are administrated in vivo they migrate to damaged tissues to replace defective cells and they can also induce peripheral tolerance [13]. In fact, MSCs reduce graft versus-host disease (GVHD) by regulating T cells, B cells, natural killer cells, monocytes, and dendritic cells [14]. The ability of MSCs to migrate is due to several chemokines and their related receptors such as PDGF (platelet-derived growth factor)/PDGF receptor, SDF-1 (stromal cell-derived factor 1)/ CXCR4(CXC chemokine receptor type, 4) axis, SCF (stem cell factor) /c-kit axis, VEGF (vascular endothelial growth factor/VEGF receptor, MCP-1(monocyte chemo-attractant protein-1)/C-C chemokine receptor type 2 [15]. Furthermore, MSC can be modified to carry therapeutic genes, serving as programmed molecule transmitters [16]. All these features make MSCs appealing for a therapeutic use in pathologies involving bone defects. Still much research needs to be done: their potential for bone regeneration is clear but the focus is on how to use MSC clinically. In order to reach this goal a thorough understanding of the pathways involved in stem cells differentiation is essential.
MSCs and HPP
Several clinical trials have reported an improvement of skeletal mineralization following bone marrow transplantation (BMT) in genetic disorders such as osteogenesis imperfecta [17]. Positive effects have been demonstrated also in HPP patients receiving bone marrow cells [18]. The beneficial effects of BMT are ascribable to the presence of MSCs and their inherent multipotency. BMT, as well as infusions with BM stromal cells or cultured osteoblasts performed in HPP patients, nevertheless, showed limited efficacy, while there was no direct evidence that these procedures led to new bone formation [19,20]. In particular, Cahill et al transplanted bone fragments and cultured osteoblasts but less than 1% of allogeneic cells were found in HPP bone 20 months after transplantation [19].
Recent approaches have suggested that BMT in combination with additional infusions of in vitro expanded MSCs improved patients' conditions as well as skeletal mineralization. Taketani et al. reported an unfortunate case where a combination of MSC with Fludarabine/cyclophosphamide used for the treatment of HPP caused leukemia with minor BCR-ABL (Ph+) in a 32-month-old girl. The authors yet suggested that alkaline phosphatase deficiency per se was not responsible for the malignancy [21]. Another study reported a complex approach consisting of expanded allogeneic MSCs, infused jointly with in vitro MSC-derived osteoblasts [22]. The authors observed poor bone mineralization and persistent low TNSALP serum levels. Recently, the same authors have used a different approach in two patients with severe HPP by combining BMT and ex vivo expanded MSCs (MSCT) [23]. The donors were first- or second-degree relatives with normal serum TNSALP levels and no mutations in ALPL gene. The clinical conditions improved in both patients after a single allogeneic BMT followed by multiple allogeneic MSCT. Bone mineralization as well as muscle mass increased in both patients although serum TNSALP levels did not normalize. Their respiratory capacity increased and their psychophysical development improved. The development of bone architecture was nevertheless abnormal.
Katsube et al. devised an intriguing approach to restore HPP MSCs functions by means of gene therapy [24]. The authors transduced a vector containing the promoter-driven ALPL gene in bone marrow-MSCs of a HPP patient. The restoration of cellular functions was demonstrated by transplantation of the engineered MSCs in nude rats. In rats the cells survived in subcutaneous sites for at least 6 weeks and were able to differentiate into osteoblasts, hence producing new bone. Further studies need to be performed in order to evaluate the efficacy of this promising therapeutic approach, which configures the possibility to employ genetically modified autologous MSCs for the treatment of HPP or other genetic skeletal diseases.
Conclusion
The therapeutic promise of MSCs for bone-related disorders is rising [25] but, in spite of the benefits associated to MSCT so far reported, further studies are needed to evaluate the molecular processes involved in MSCs engraftment and their osteogenic differentiation in HPP patients. Understanding the mechanisms through which MSCT acts in these patients can help to identify the most effective approach suitable to restore bone architecture and functions not only for HPP but also for other skeletal related diseases.
References
-
Mornet E (2007) Hypophosphatasia. Orphanet J Rare Dis 2: 40.
-
Millán JL, Plotkin H (2012) Hypophosphatasia - pathophysiology and treatment. Actual osteol 8: 164-182.
-
Whyte MP, Greenberg CR, Salman NJ, Bober MB, McAlister WH, et al. (2012) Enzyme-replacement therapy in life-threatening hypophosphatasia. N Engl J Med 366: 904-913.
-
Matsumoto T, Miyake K, Yamamoto S, Orimo H, Miyake N, et al. (2011) Rescue of severe infantile hypophosphatasia mice by AAV-mediated sustained expression of soluble alkaline phosphatase. Human gene therapy 22: 1355-1364.
-
Kosnik-Infinger L, Gendron C, Gordon CB, Pan BS, van Aalst JA, et al. (2015) Enzyme replacement therapy for congenital hypophosphatasia allows for surgical treatment of related complex craniosynostosis: a case series. Neurosurg Focus 38: E10.
-
Bianchi ML (2015) Hypophosphatasia: an overview of the disease and its treatment. Osteoporosis international: a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA 26: 2743-2757.
-
Hessle L, Johnson KA, Anderson HC, Narisawa S, Sali A, et al. (2002) Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proceedings of the National Academy of Sciences of the United States of America 99: 9445-9449.
-
Wang X, Wang Y, Gou W, Lu Q, Peng J, et al. (2013) Role of mesenchymal stem cells in bone regeneration and fracture repair: a review. Int Orthop 37: 2491-2498.
-
Ivkovic A, Marijanovic I, Hudetz D, Porter RM, Pecina M, et al. (2011) Regenerative medicine and tissue engineering in orthopaedic surgery. Front Biosci (Elite Ed) 3: 923-944.
-
Jethva R, Otsuru S, Dominici M, Horwitz EM (2009) Cell therapy for disorders of bone. Cytotherapy 11: 3-17.
-
Le Blanc K, Pittenger M (2005) Mesenchymal stem cells: progress toward promise. Cytotherapy 7: 36-45.
-
Valenti MT, Dalle Carbonare L, Donatelli L, Bertoldo F, Zanatta M, et al. (2008) Gene expression analysis in osteoblastic differentiation from peripheral blood mesenchymal stem cells. Bone 43: 1084-1092.
-
Desiderio V, Tirino V, Papaccio G, Paino F (2014) Bone defects: molecular and cellular therapeutic targets. Int J Biochem Cell Biol 51: 75-78.
-
Abdallah BM, Kassem M (2009) The use of mesenchymal (skeletal) stem cells for treatment of degenerative diseases: current status and future perspectives. Journal of cellular physiology 218: 9-12.
-
Liu Y, Wu J, Zhu Y, Han J (2014) Therapeutic application of mesenchymal stem cells in bone and joint diseases. Clin Exp Med 14: 13-24.
-
Lu CH, Chang YH, Lin SY, Li KC, Hu YC (2013) Recent progresses in gene delivery-based bone tissue engineering. Biotechnol Adv 31: 1695-1706.
-
Horwitz EM, Gordon PL, Koo WKK, Marx JC, Neel MD, et al. (2002) Isolated allogeneic bone marrow-derived mesenchymal cells engraft and stimulate growth in children with osteogenesis imperfecta: Implications for cell therapy of bone. Proceedings of the National Academy of Sciences of the United States of America 99: 8932-8937.
-
Cahill RA, Jones OY, Klemperer M, Steele A, Mueller TO, et al. (2004) Replacement of recipient stromal/mesenchymal cells after bone marrow transplantation using bone fragments and cultured osteoblast-like cells. Biology of blood and marrow transplantation: journal of the American Society for Blood and Marrow Transplantation 10: 709-717.
-
Cahill RA, Wenkert D, Perlman SA, Steele A, Coburn SP, et al. (2007) Infantile hypophosphatasia: transplantation therapy trial using bone fragments and cultured osteoblasts. The Journal of clinical endocrinology and metabolism 92: 2923-2930
-
Whyte MP, Kurtzberg J, McAlister WH, Mumm S, Podgornik MN, et al. (2003) Marrow cell transplantation for infantile hypophosphatasia. J Bone Miner Res 18: 624-636.
-
Taketani T, Kanai R, Abe M, Mishima S, Tadokoro M, et al. (2013) Therapy-related Ph+ leukemia after both bone marrow and mesenchymal stem cell transplantation for hypophosphatasia. Pediatr Int 55: e52-55.
-
Tadokoro M, Kanai R, Taketani T, Uchio Y, Yamaguchi S, et al. (2009) New bone formation by allogeneic mesenchymal stem cell transplantation in a patient with perinatal hypophosphatasia. J Pediatr 154: 924-930.
-
Taketani T, Oyama C, Mihara A, Tanabe Y, Abe M, et al. (2015) Ex Vivo Expanded Allogeneic Mesenchymal Stem Cells With Bone Marrow Transplantation Improved Osteogenesis in Infants With Severe Hypophosphatasia. Cell transplantation 24: 1931-1943.
-
Katsube Y, Kotobuki N, Tadokoro M, Kanai R, Taketani T, et al. (2010) Restoration of cellular function of mesenchymal stem cells from a hypophosphatasia patient. Gene Ther 17: 494-502.
-
D'souza N, Rossignoli F, Golinelli G, Grisendi G, Spano C, et al. (2015) Mesenchymal stem/stromal cells as a delivery platform in cell and gene therapies. BMC Med 13: 186.
