

Journal of Genetics and Genome Research
Discordant Disease Course in a Monozygotic Twin Pair with Juvenile Myelomonocytic Leukemia
Silvia Bresolin1*, Paola De Filippi3, Claudia Cagioni3, Simone Cesaro4, Annamaria Di Meglio2, Anna Leszl1, Fiorenza Aprili5, Chiara Cugno6, M.D., Chiara Frasson2, Marco Zecca6, Cesare Danesino7,8 and Geertruyte Kronnie1
1Department of Women's and Children's Health, University of Padova, Padova, Italy
2Department of Women's and Children's Health, University of Padova, Istituto di Ricerca, Pediatrica Città della Speranza, Padova, Italy
3Department of Molecular Medicine, University of Pavia, Pavia, Italy
4Pediatric Hematology Oncology, Azienda Ospedaliera Universitaria Integrata, Verona, Italy
5Clinical Chemistry Laboratory, Azienda Ospedaliera Universitaria Integrata, Verona, Italy
6Pediatric Hematology-Oncology, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy
7Department of Molecular Medicine, University of Pavia, Pavia, Italy
8Department of Molecular Medicine, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy
*Corresponding author:
Bresolin Silvia, Laboratory of Oncohematology, Department of Women's and Children's Health, University of Padova, Padova, Italy, Tel: +39 049 8211453; E-mail: silvia.bresolin@unipd.it
J Genet Genome Res, JGGR-1-009, (Volume 1, Issue 2), Research Article; ISSN: 2378-3648
Received: November 14, 2014 | Accepted: December 10, 2014 | Published: December 14, 2014
Citation: Bresolin S, Filippi PD, Cagioni C, Cesaro S, Meglio AD, et al. (2014) Discordant Disease Course in a Monozygotic Twin Pair with Juvenile Myelomonocytic Leukemia. J Genet Genome Res 1:009. 10.23937/2378-3648/1410009
Copyright: © 2014 Bresolin S, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Juvenile myelomonocytic leukemia is a rare neoplastic disorder occurring in early childhood often showing an aggressive progression. We report a case of a twin pair with concordant JMML but an extremely different disease course. Both twins presented with somatic aberrations of chromosome 7 and mutations in PTPN11. Analysis of sorted BM and PB cell populations revealed the clonal nature of the disease and indicated that genomic aberrations arise from common hematopoietic precursor cells. PTPN11 mutations in oral swab specimens of both patients during the active phase of the disease were attributed to monocytes infiltrating the oral mucosa and not to the presence of mosaic tissue mutations. This study provides evidence that the discordant clinical disease course in the twins is associated with a distinct gene expression profile.
Keywords
Juvenile myelomonocytic leukemia, Monozygotic twins, Discordant disease
Introduction
Juvenile myelomonocytic leukemia (JMML) is a rare myelodysplastic/myeloproliferative disorder, characterized by the malignant transformation of myeloid progenitors in the stem cell compartment [1]. Clinical features like hepatosplenomegaly, lymphadenopathy, pallor, fever and skin rash, peripheral blood monocyte counts >1x109/L, bone marrow blast counts < 20% and the absence of BCR-ABL1 rearrangements are usually indicative of JMML [2], yet diagnosis can be established with certainty only after laboratory workup has confirmed the presence of clonal abnormalities, including monosomy 7 and/or point mutations resulting in the activation of the RAS/MAPK cascade [3].
Leukemia is not generally considered to be inherited but its incidence in twins is high and this has been related to a common clonal origin of the disease. Generally, accepted basis of concordance of leukemia is that following the genomic alterations in one twin fetus, these aberrations spread to the co-twin via a common monochorionic placenta [4]. Seventyfive percent of monozygotic twins are monochorionic hence presenting a high probability of blood cell exchange [5,6].
Clinicians and parents facing a severely diseased child of a twin pair are frequently concerned about concordance of the disorder even in the absence in the co-twin of specific clinical features or any clear evidence as to the constitutional genetic background of the disease.
The clonal origin of JMML has been demonstrated in patients with RAS mutations [7] and NF1 deletions [8]. However, cell differentiation stages where aberrations occur are not univocal. As to JMML patients with RAS mutations, one patient presented aberrations in both the myeloid lineage and the B-lineage, whereas the mutations were restricted only to the myeloid lineage in two other patients [7]. One case of JMML with NF1 harbored the mutation in the myeloid population and in the derived T-cell lymphoma, suggesting the pluripotent stem cell as the cell of origin [8].
In a murine model of myeloproliferative disease a conditional knock-in mutation of Ptpn11E76K in pan hematopoietic cells resulted in the activation of hematopoietic stem cells (HSCs) and myeloid progenitors [9].
With respect to the timing of appearance of the mutation during hematopoietic stem cell differentiation, no data are currently available for JMML patients with the most common PTPN11 mutation (i.e. 35% of patients).
This report presents the results of a two-year study on a monozygotic twin pair suffering from concordant juvenile myelomonocytic leukemia (JMML), carrying the same PTPN11 mutation and a rare 46, XY, -7 +mar karyotype but presenting a very different disease course. A clinical case report described the outcome of allogeneic hematopoietic stem cell transplant in this twin pair [10].
Methods
Materials and methods are described in full detail in the Supplemental Appendix.
In summary, we used the following approaches to analyze the specimens of a twin pair with JMML: microarray gene expression profiling and classification of the diagnostic samples, using the diagnostic classifier model [11]; cytogenetics, including karyotyping, FISH and aCGH to define the specific chromosome 7 aberration at relapse of Twin_01; flow cytometric sorting of whole bone marrow and peripheral blood in subpopulations, according to cell lineage origin and differentiation state; Sanger sequencing and Amplicon Ultra Deep 454 sequencing of PTPN11 mutations to determine mutant allele frequencies; human leukocyte antigen (HLA) and short tandem repeats (STR) analyses of monozygosity; specific STR analysis of chromosome 7 to determine origin and extent of chromosome 7 aberrations.
Results and Discussion
A twin pair with concordant JMML
This study refers to a pair of monozygotic, monochorionic twins born at 36 weeks of gestation. At first presentation (age 19 month) Twin_01 was suffering from fever, low platelet count, anemia and hepatosplenomegaly. Peripheral blood (PB) smears showed monocytosis and low blast count (3% of blasts). A few months later, the bone marrow specimen showed dysplasia in myeloid cell lineages. The co-twin (Twin_02) presented no clinical symptoms of JMML at the same age but laboratory analysis revealed blood morphology and blood count data compatible with the diagnosis of JMML. Symptoms appeared 4 months later.
Analysis of the HLA system and STR analysis confirmed the monozygotic origin of the twins (Supplemental Figure 1). According to differential diagnostic criteria, each twin received a diagnosis of JMML with a common PTPN11 mutation and abnormal chromosome 7 [10]. Disease timeline and progression is shown in Figure 1, whereas treatment details were reported earlier [10]
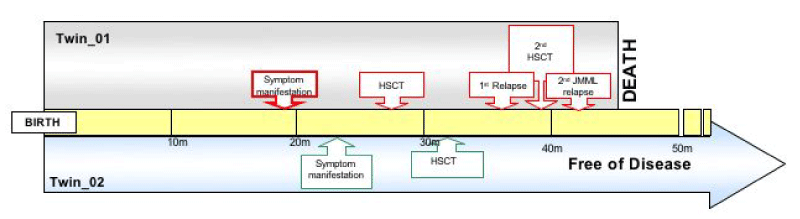 Figure 1: Timeline of the disease course in both twins. Twin_01 timeline from birth to death at 45 months. Twin_02 timeline from birth to follow-up at 48 months, in complete remission.
View Figure 1
Figure 1: Timeline of the disease course in both twins. Twin_01 timeline from birth to death at 45 months. Twin_02 timeline from birth to follow-up at 48 months, in complete remission.
View Figure 1
 Supplemental Figure 1: Short tandem repeats (STR) profiles of both twins in fibroblast cells. STR approach for forensic human identification was used to identify with high accuracy the monozygosity of the twins. Identical profiles were obtained from both twins.
View Supplemental Figure 1
Supplemental Figure 1: Short tandem repeats (STR) profiles of both twins in fibroblast cells. STR approach for forensic human identification was used to identify with high accuracy the monozygosity of the twins. Identical profiles were obtained from both twins.
View Supplemental Figure 1
Clonal origin of JMML in twins with PTPN11 mutation and aberrant chromosome 7
Historical records on monozygotic twins with concordant leukemia revealed a high probability of a common origin for the disease, leukemia stem cells in one twin fetus being transmitted to the co-twin via a common placenta [4]. Table 1 shows 6 cases of concordant JMML in monozygotic twins, suggesting a common clonal origin of the disease, even though, in most cases, no information concerning chorionic/amniotic type and genomic aberrations is provided [12-17]. In addition, absence of disease concordance has also been noted in at least one case of JMML (Table 1) [18].
![]()
Table 1: Historic record of JMML (previously named JCML or jCMML) twin cases.
View Table 1
We present here the case of a twin pair with confirmed monozygosity and monochorionic placentation, a PTPN11E76K mutation and a karyotype with -7 mar+. Array CGH had delineated diploid chromosome 7, p12.1 harboring sequences of 3 genes and known noncoding RNAs in each of the twins [10]. The presence of the same rare aberration of der 7 of maternal origin along with the same mutations of PTPN11 strongly corroborates the hypothesis that both events occurred in utero in one twin fetus first, the aberrations later being transmitted to the co-twin via the common placenta. Previous detection of KRAS and PTPN11 mutations in Guthrie cards of JMML patients is in line with their prenatal occurrence [19]. The possibility that the above aberrations in the twins could have occurred in utero as unrelated events is refuted.
Non-lineage restriction of genetic JMML markersThe clonal origin of JMML has long been recognized [7,8], but the involvement of all three major lineages (i.e. myeloid, B and T lymphoid) is a new finding.
The loss of both arms of chromosome 7 and the PTPN11 mutation in all subpopulations of the myeloid lineage as well as in the B and T-lineage show that both JMML inciting events occurred in the common hematopoietic precursors (Supplemental Figure 2, Supplemental Table 1). We speculate that these common hematopoietic stem cells resided in the hypoxic BM niche of Twin_01 and that slightly differentiated progenitors left the niche and reached Twin_02 through common placental circulation.
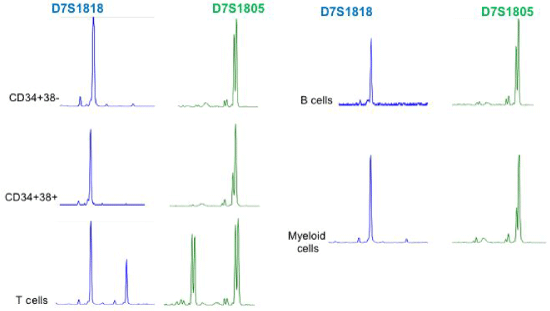 Supplemental Figure 2: Short Tandem Repeat analysis to detect monosomy 7 in subpopulations of total bone marrow in Twin_02 Detection of monosomy 7 was performed using two markers of chromosome 7, i.e. D7S1818 and D7S1805. In distinct subpopulations, loss to various extent of material of chromosome 7 was revealed. Twin_01 showed similar losses of chromosome 7 in subpopulations at diagnosis and the same losses were revealed at relapse.
View Supplemental Figure 2
Supplemental Figure 2: Short Tandem Repeat analysis to detect monosomy 7 in subpopulations of total bone marrow in Twin_02 Detection of monosomy 7 was performed using two markers of chromosome 7, i.e. D7S1818 and D7S1805. In distinct subpopulations, loss to various extent of material of chromosome 7 was revealed. Twin_01 showed similar losses of chromosome 7 in subpopulations at diagnosis and the same losses were revealed at relapse.
View Supplemental Figure 2
Mutation load of PTPN11 in whole BM and PB samples diverged from the maximum of 50% for heterozygous mutations revealing a considerable presence of cells without PTPN11 mutations Supplemental Appendix.; similarly, the FISH analysis had shown the presence of nuclei with a normal karyotype. Comparable results were found in all cell subpopulations, which may suggest that also normal cells are present in the hematopoietic niche of the BM. The high percentage of non-mutated cells observed in PB samples may indicate that mutated hematopoietic cells are extremely prone to infiltrate different tissues.
Distinct gene expression signatures in Twin_01 and Twin_02
The twin pair showed two distinct gene expression signatures: the diagnostic classifier revealed an AML-like signature in Twin_01 and a non AML-like signature in Twin_02 (Supplemental Table 2, Supplemental Appendix.). Remarkably, Twin_01 relapsed twice following HSCT and deceased, whereas Twin_02 remained in remission after HSCT.
The highly discordant clinical disease course in the twins (Figure 1) is in close concordance with our results on 44 cases with JMML. This study showing that patients with an AML-like signature have a poor prognosis, whereas patients with an non-AML-like signature have a favorable prognosis following HSCT [11]. The co-occurrence of monosomy 7 and PTPN11 mutation in the twins with an AML-like and a non-AML-like signature was also previously found to be present in patients with distinct GEP-based classification [11].
As described at diagnosis, the BM of Twin_01 presented with the same PTPN11E67K mutation, as well as the loss of the long and short arms of chromosome 7, der7, p12.1 and an AML-like GEP signature, also at relapse (Supplemental Figure 3).
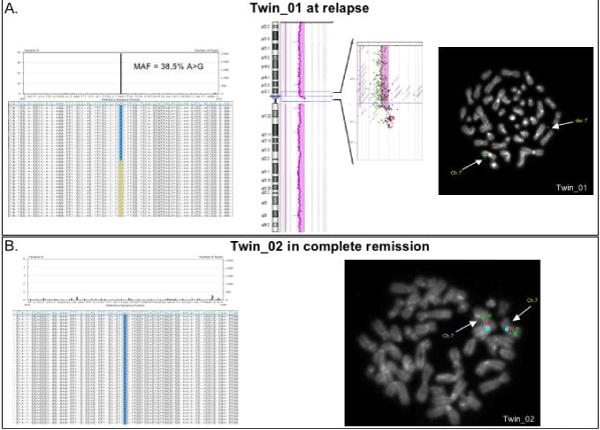 Supplemental Figure 3: Molecular features of the twins after HSCT.
Supplemental Figure 3: Molecular features of the twins after HSCT.
A. Twin_01 at relapse. The molecular features of Twin_01 found at diagnosis remained unchanged at relapse. Ultra deep sequencing confirmed the same PTPN11 mutation at (E76K) and FISH analyses revealed the presence of der 7 with the conserved p12.1 region, including 41 transcripts (array CGH profile, in the middle). On the top, left-hand side of the figure, we show reads obtained with 454 for BM of Twin_01 at relapse. Substitution of A in G was identified with a MAF of 38,5%. B. Twin_02 in complete remission. Twin_02 ultra deep sequencing of PTPN11 mutation in exon 3 and FISH analysis showed normal features of the hematopoietic cells. On the bottom, left-hand side of the figure, we show reads obtained with 454 for BM of Twin_02 in complete remission after HSCT; no mutated reads were obtained compared to reference sequence of exon 3.
FISH analysis in both twins (right-hand side of the figure) was performed using probe CEP 7 Spectrum Aqua (Blue: B); Williams Region Probe FISH Probe Kit: ELN (7q11.23), Spectrum Orange (Red: R) / D7S486, D7S522 (7q31) Spectrum Green (Green: G). In the metaphase, chromosome 7 (signals B,R,G), and "der 7" (smaller signal) were identified, as indicated by the arrows.
View Supplemental Figure 3
Mosaic tissue mutations vs. disease related infiltration with mutated leukocytes
Constitutional mutation syndromes have been related to JMML along with recurrent mutations including NF1 (Neurofibromatosis type 1); PTPN11, KRAS, NRAS (Noonan syndrome) and CBL [20-22]. Also mosaicism in non-hematopoietic tissues for KRAS and NRAS has been recently reported in JMML patients [23]. Even though Noonan syndrome phenotypical features were absent in the twins, we analyzed other tissues looking for PTPN11 mutations in order to rule out the hypothesis that the genomic aberrations in the common hematopoietic progenitor cells may have occurred also in common mesentoderm progenitor cells.
We analyzed fibroblasts, hair follicles and oral swabs to fully exclude the presence of mutations beyond the hematopoietic compartment. No mutation of PTPN11 was detected in either fibroblasts or hair bulbs. We did find PTPN11 mutations in oral swab during the active phase of the disease (Supplemental Figure 4) but not following HSCT, when the twins were in remission. We assume that the PTPN11 mutations in both patients during the active phase of the disease with very similar mutant allele frequency can be attributed to monocytes infiltrating the oral mucosa and not to the presence of mosaic tissue mutations [23] (Supplemental Figure 5).
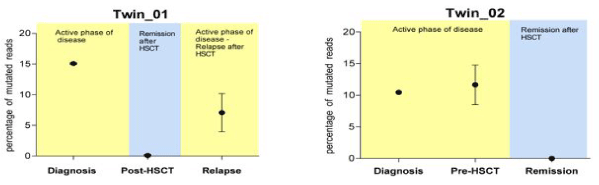 Supplemental Figure 4: PTPN11 mutations in oral swabs in the twins. Using 454 technologies, the presence of PTPN11 mutations in the oral swabs was detected only in the active phase of the disease (for Twin_01 at diagnosis and relapse and at diagnosis only in Twin_02). No PTPN11 mutations were identified after HSCT or during the remission phase. On the x-axis, oral swabs at different time points are indicated, i.e. at diagnosis, pre-HSCT, post-HSCT and at relapse after HSCT. On the y-axis, the percentage of mutated reads identified using 454 technology is indicated. Each point gives the mean values of three independent oral swabs.
View Supplemental Figure 4
Supplemental Figure 4: PTPN11 mutations in oral swabs in the twins. Using 454 technologies, the presence of PTPN11 mutations in the oral swabs was detected only in the active phase of the disease (for Twin_01 at diagnosis and relapse and at diagnosis only in Twin_02). No PTPN11 mutations were identified after HSCT or during the remission phase. On the x-axis, oral swabs at different time points are indicated, i.e. at diagnosis, pre-HSCT, post-HSCT and at relapse after HSCT. On the y-axis, the percentage of mutated reads identified using 454 technology is indicated. Each point gives the mean values of three independent oral swabs.
View Supplemental Figure 4
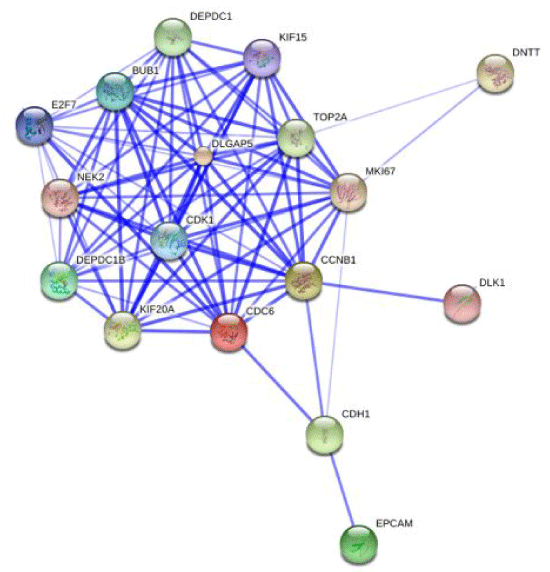 Supplemental Figure 5: Network diagram of protein and their interactions. Representative protein-protein interaction network of cell cycle enriched genes differentially expressed in Twin_01 and Twin_02 generated from STRING database. The connecting lines indicate functional relationships and direct protein-protein interactions.
View Supplemental Figure 5
Supplemental Figure 5: Network diagram of protein and their interactions. Representative protein-protein interaction network of cell cycle enriched genes differentially expressed in Twin_01 and Twin_02 generated from STRING database. The connecting lines indicate functional relationships and direct protein-protein interactions.
View Supplemental Figure 5
With respect to the absence of PTPN11 mutations, also two copies of chromosome 7 were found in fibroblasts, hair bulbs and during remission in oral swabs. Aberrations of chromosome 7 were detected only in the active phase of the disease.
Implications for clinicians and translational research
Our study of a twin pair with JMML and identical genetic and karyotypic features and historical reports of JMML in twins, leads us to infer that the disease triggering mutations were already present at birth in each monozygotic twin. To the best of our knowledge, only one monochorionic-diamniotic twin pair has been described so far and only one of the twins suffered from JMML [18]. This condition may be due to the absence of transfer of the NRAS mutated clone from one twin to the other during prenatal life.
In the case reported here, the seeding of cells with both inciting genetic aberrations in the twin pair occurred long before the onset of the disease at the age of two, as inferred from the common prenatal origin. Also the presence of the mutations was demonstrated in Twin_02 before the disease became clinically manifest. For this reason, along with historical data (Table 1) pointing to extremely high concordance of JMML in twins, our data indicate that an immediate diagnostic workup for differential diagnosis of JMML in twins is mandatory, in case one of them presents with clinical features suggestive of JMML.
Mutant allele frequency levels in total BM and PB specimens and in the sorted cell populations (Supplemental Appendix., Supplemental Table 1) also showed that nonmutated cells can be found at all levels of differentiation. This suggests that there is still room for investigation into the therapeutic strategies aiming to spare healthy cells and eliminate mutated cells.
Monocytic leukemias are highly invasive and spread to other tissues, especially to the spleen, skin and mucosa. As a consequence, oral swabs taken in the active phase of the disease as well as other tissues that are subject to monocyte infiltration are not the most appropriate specimens, if we aim to look for any constitutional or mosaic tissue mutations, since the mutations may be attributed to infiltrating mutated leucocytes. In case of positive oral swab specimens it is advisable to repeat the mutation analysis on different tissues.
Finally, non-AML-like signatures in one of the twins with JMML warrants success of current therapies, whereas AML-like signatures along with a high incidence of relapses highlight the need to eliminate the mutated common hematopoietic cells in the bone marrow niche.
Acknowledgments
We thank Svetlana Donska, M.D, Pediatric Hematology Oncology, Children Hematology Center, Okhmatdet Hospital, Kiev, Ukraine, for referring the twins to SC and Lifeline Italia Trust for the unrestricted economical support to the family. This work was partly supported by grants from AIRC (S.B.), "Fondazione Sofia Luce Rebuffat" (M.Z.) and Fondazione Città della Speranza (C.F., A.dM.).
References
-
Niemeyer CM, Arico M, Basso G, Biondi A, Cantu Rajnoldi A, et al. (1997) Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. European Working Group on Myelodysplastic Syndromes in Childhood (EWOG-MDS) Blood 89: 3534-3543.
-
Hasle H, Niemeyer CM, Chessells JM, Baumann I, Bennett JM, et al. (2003) A pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases. Leukemia 17: 277-282.
-
Niemeyer CM, Kratz CP (2008) Paediatric myelodysplastic syndromes and juvenile myelomonocytic leukaemia: molecular classification and treatment options. Br J Haematol 140: 610-624.
-
Greaves MF, Maia AT, Wiemels JL, Ford AM (2003) Leukemia in twins: lessons in natural history. Blood 102: 2321-2333.
-
Falletta JM, Starling KA, Fernbach DJ (1973) Leukemia in twins. Pediatrics 52: 846-849.
-
Wiemels JL, Cazzaniga G, Daniotti M, Eden OB, Addison GM, et al. (1999) Prenatal origin of acute lymphoblastic leukaemia in children. Lancet 354: 1499-1503.
-
Flotho C, Valcamonica S, Mach-Pascual S, Schmahl G, Corral L, et al. (1999) RAS mutations and clonality analysis in children with juvenile myelomonocytic leukemia (JMML). Leukemia 13: 32-37.
-
Cooper LJ, Shannon KM, Loken MR, Weaver M, Stephens K, et al. (2000) Evidence that juvenile myelomonocytic leukemia can arise from a pluripotential stem cell. Blood 96: 2310-2313.
-
Xu D, Liu X, Yu WM, Meyerson HJ, Guo C, et al. (2011) Non-lineage/stage-restricted effects of a gain-of-function mutation in tyrosine phosphatase Ptpn11 (Shp2) on malignant transformation of hematopoietic cells. J Exp Med 208: 1977-1988.
-
Cesaro S, De Filippi P, Di Meglio A, Leszl A, Donska S, et al. (2014) Different outcomes of allogeneic hematopoietic stem cell transplant in a pair of twins affected by juvenile myelomonocytic leukemia. Int J Hematol 99: 208-212.
-
Bresolin S, Zecca M, Flotho C, Trentin L, Zangrando A, et al. (2010) Gene expression-based classification as an independent predictor of clinical outcome in juvenile myelomonocytic leukemia. J Clin Oncol 28: 1919-1927.
-
12. Svarch E, de la Torre E (1977) Myelomonocytic leukaemia with a preleukaemic syndrome and Ph1 chromosome in monozygotic twins. Arch Dis Child 52: 72-74.
-
Castro-Malaspina H, Schaison G, Passe S, Pasquier A, Berger R, et al. (1984) Subacute and chronic myelomonocytic leukemia in children (juvenile CML). Clinical and hematologic observations, and identification of prognostic factors. Cancer 54: 675-686.
-
Massaad L, Prieur M, Gaud C, Fischer A, Dutrillaux B (1989) Unusual karyotypic evolution in subacute myelomonocytic leukemia in two monozygotic twins. Cancer Genet Cytogenet 38: 205-213.
-
Passmore SJ, Hann IM, Stiller CA, Ramani P, Swansbury GJ, et al. (1995) Pediatric myelodysplasia: a study of 68 children and a new prognostic scoring system. Blood 85: 1742-1750.
-
Najfeld V, Vlachos A, Parker R, Burnett W, Scalise A, et al. (1997) Evidence for the embryonic origin of partial chromosome 7 deletion in monozygotic twins with juvenile chronic myelogenous leukemia. Leukemia 11: 306-310.
-
Mark Z, Toren A, Amariglio N, Vonsover A, Rechavi G, et al. (1995) Rearrangement of the immunoglobulin heavy chain gene in juvenile chronic myeloid leukaemia. Br J Haematol 90: 353-357.
-
Tanoshima R, Goto H, Yanagimachi M, Kajiwara R, Kuroki F, et al. (2008) Graft versus leukemia effect against juvenile myelomonocytic leukemia after unrelated cord blood transplantation. Pediatr Blood Cancer 50: 665-667.
-
Matsuda K, Sakashita K, Taira C, Tanaka-Yanagisawa M, Yanagisawa R, et al. (2010) Quantitative assessment of PTPN11 or RAS mutations at the neonatal period and during the clinical course in patients with juvenile myelomonocytic leukaemia. Br J Haematol 148: 593-599.
-
Shannon KM, O'Connell P, Martin GA, Paderanga D, Olson K, et al. (1994) Loss of the normal NF1 allele from the bone marrow of children with type 1 neurofibromatosis and malignant myeloid disorders. N Engl J Med 330: 597-601.
-
Flotho C, Steinemann D, Mullighan CG, Neale G, Mayer K, et al. (2007) Genome-wide single-nucleotide polymorphism analysis in juvenile myelomonocytic leukemia identifies uniparental disomy surrounding the NF1 locus in cases associated with neurofibromatosis but not in cases with mutant RAS or PTPN11. Oncogene 26: 5816-5821.
-
Pérez B, Mechinaud F, Galambrun C, Ben Romdhane N, Isidor B, et al. (2010) Germline mutations of the CBL gene define a new genetic syndrome with predisposition to juvenile myelomonocytic leukaemia. J Med Genet 47: 686-691.
-
Doisaki S, Muramatsu H, Shimada A, Takahashi Y, Mori-Ezaki M, et al. (2012) Somatic mosaicism for oncogenic NRAS mutations in juvenile myelomonocytic leukemia. Blood 120: 1485-1488.
