

Journal of Genetics and Genome Research
Enhanced Detection of Longer Insertions and Deletions in Clinical Exome Sequencing Improves Diagnostic Yield
Deepali N. Shinde*, Jefferey Chen, Soren Fischbach, David J. Salvador, Kelly Farwell, Hsiao-Mei Lu and Sha Tang
Ambry Genetics, 15 Argonaut, Aliso Viejo, CA, USA
*Corresponding author:
Deepali N. Shinde, Ambry Genetics, USA, E-mail: dshinde@ambrygen.com
J Genet Genome Res, JGGR-2-018, (Volume 2, Issue 2), Research Article; ISSN: 2378-3648
Received: May 17, 2015 | Accepted: September 14, 2015 | Published: September 17, 2015
Citation: Shinde DN, Chen J, Fischbach S, Salvador DJ, Farwell K, et al. (2015) Enhanced Detection of Longer Insertions and Deletions in Clinical Exome Sequencing Improves Diagnostic Yield. J Genet Genome Res 2:018. 10.23937/2378-3648/1410018
Copyright: © 2015 Shinde DN, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Whole exome sequencing (WES) has been remarkably successful as both a diagnostic and novel gene discovery tool since its introduction to the clinical laboratory in 2011. Where traditional diagnostic methods have been uninformative in discovering the pathogenic etiology in patients, diagnostic exome sequencing (DES) has provided answers for roughly one-third of patients tested, thus contributing to the management of patients' overall healthcare. Single nucleotide variants are generally efficiently identified by DES in well-covered exonic regions. However, accurate mapping of insertions and deletions, especially those larger than 20 nucleotides, is challenging due to gapped alignment and paired-end sequence inference. We have customized and validated a robust exome analysis pipeline that accurately and efficiently calls insertions or deletions ranging from 20 to 200 base pairs from next generation sequencing data and contributes to one of the highest diagnostic yields reported for clinical exome analysis. Out of 284 positive/likely positive cases in the first 1000 unselected DES cases referred to Ambry Genetics, causative mutations in 9 (3.2%) were associated with insertions, deletions or indels between 20 and 200 bp in length. Our data highlight the importance of an optimized clinical exome workflow for the detection of longer insertions and deletions to improve clinical sensitivity and diagnostic yield.
Keywords
Insertions, Deletions, INDEL, Diagnostic exome sequencing, DES, Whole exome sequencing, WES, Next generation sequencing, NGS, Bioinformatics
Introduction
An ability to simultaneously and rapidly investigate variants in about 20,000 genes and continually decreasing costs have promoted the growing use of next generation sequencing (NGS)-based technologies such as whole exome sequencing (WES) in clinical diagnostics for personalized medicine (reviewed in [1]). However, laboratories conducting clinical WES have been able to achieve at best a 25-30% diagnostic rate [2-4] despite the estimates that 85% of human Mendelian diseases are caused by mutations in exons [5]. There is clearly a need for improvement in variant detection within these exons and the challenge now is the computational analysis of the ever-increasing amount of data generated by WES and the requirement for a robust bioinformatics pipeline for sequence alignment to a reference genome, variant calling, annotation, filtering and variant prioritization [6]. Multiple commercial and freely available software programs have been developed for each of these steps and rigorous validation of the pipeline integrating one or more of these programs can increase the efficiency and sensitivity of variant detection from patient samples [7].
Two types of DNA sequence alterations are reported following diagnostic exome sequencing (DES) - single nucleotide variants (SNVs) and insertions, deletions or indels (co-localized insertions and deletions; collectively referred to as indels for the purpose of simplicity). SNVs are the most common type of genetic alterations found in the human genome and substantial amount of research, such as that by the International HapMap Consortium, has been focused on accurately mapping and identifying SNVs for human genetic variation studies [8]. However, in spite of being the second most common type of genomic alterations [9], indels are more challenging to identify due to sequence alignment issues which can be complicated by the presence of single nucleotide polymorphisms or sequencing errors that prevent perfect ungapped alignment with the reference genome [10]. In addition, differences in sizes of the relevant coding exons, coverage in terms of the numbers of reads and read lengths shorter than the length of the indel have limited the reporting of indels larger than 15 nucleotides [11]. Here we report the development and application of an optimized exome pipeline that was successfully used to identify indels more than 20 nucleotides in length from patients' samples and provide answers to long and uninformative diagnostic odysseys in order to aid their overall healthcare management.
Terminology
WES: Whole Exome sequencing; sequencing of almost all coding exons in the genome.
DES: Diagnostic Exome sequencing; WES performed for molecular diagnostics in a single patient.
Variant calling Q-score: For each SNP or indel call, the probability of both the called genotype and any non-reference genotype is provided as a quality score (Q-score).
CASAVA: CASAVA (short for "Consensus Assessment of Sequence and Variation") is the part of Illumina's sequencing analysis software that performs alignment of a sequencing run to a reference genome and subsequent variant analysis and read counting.
Orphan reads: An orphan read is the unaligned part of paired reads for which only one read is aligned.
Anomalous read pairs: Anomalous read pairs contain insert sizes that are anomalously large (possible deletion) or small (possible insertion).
AVA: Ambry Variant Analyzer.
Characterized Mendelian Disease Gene (characterized gene): A gene known to underlie at least one Mendelian genetic condition.
Novel Mendelian Disease Gene (novel gene): A gene that is not currently known to underlie a Mendelian genetic condition.
Materials and Methods
Patients/study population
The first sequential 1000 patients referred to Ambry Genetics Laboratory for DES beginning in September 2011 were included in this study. When available, all first degree and affected second- or third-degree family members were included along with the proband for testing. Patient identifiers were removed. Solutions' Institutional Review Board determined the study to be exempt from the Office for Human Research Protections Regulations for the Protection of Human Subjects (45 CFR 46) under category 4. Since retrospective analysis of anonymized data was performed, the study was not required to receive consent from patients.
Whole-exome sequencing
Genomic deoxyribonucleic acid (gDNA) was isolated from whole blood from the patient and first degree relatives when available. Samples were prepared using either the SureSelect Target Enrichment System (Agilent Technologies, Santa Clara, CA) or the SeqCap EZ VCRome 2.0 (Roche NimbleGen, Madison, WI). The enriched exome libraries were sequenced using paired-end, 100-cycle chemistry on the Illumina HiSeq 2000 or 2500 (Illumina, San Diego, CA).
Bioinformatics annotation, filtering of variants, and Family history Inheritance-based Detection (FIND)
The Ambry Exome pipeline utilizes FASTQ files from CASAVA for read alignment to the human reference sequence (GRCh37), and to generate variant reports. For the detection of indels specifically, the indel caller finds indels using a two stage process during alignment, which involves identifying candidate indels in the first stage, and realigning supporting reads to each indel in the second stage. During the first stage, an indel can be identified from both gapped alignments, which efficiently identify relatively small indels (1-10 bases), and contig assembly and alignments, which identify much larger indels (> 10 bases). The two conditions that must be met when opening a gap during alignment are
1. A gap must correct at least 5 mismatches downstream (default); and
During contig assembly and alignments, non-aligned 'orphan reads' and anomalous read pairs are first clustered and assembled into contigs. The contigs are later aligned back to the reference genome. Contig assembly and alignments utilize the 'assembleINDELs' module in CASAVA that runs only on paired-end reads. During the second stage, all intersecting reads are realigned to each candidate indel and the reference genome, after which each indel's genotype and associated quality score are assigned.
In this study, variants with a variant calling Q-score of < 20 and an allele count of < 10X were filtered out and variant reports generated from the Ambry Exome pipeline were converted to an input format that was uploaded into the Ambry Variant Analyzer tool (AVA). Samples were classified into several categories and filtered out if they were located outside the analytical range of ± 2bp of the coding exons and/or determined to be a polymorphism with thresholds of 0.1% for the autosomal/X-linked dominant modes of inheritance for characterized and novel disease genes, and 1% and 0.2% for the autosomal/X-linked recessive modes of inheritance for characterized and novel disease genes, respectively (utilizing population frequency data from multiple sources including NCBI dbSNP [12], NHLBI Exome Sequencing Project (ESP), 1000 Genomes [13] and an internal Ambry database). The Human Gene Mutation Database (HGMD) [14], the Online Mendelian Inheritance in Man (OMIM) database and online search engines (e.g., PubMed) were used to search for previously described gene mutations and polymorphisms. Data were annotated with AVA, including nucleotide and amino acid conservation, biochemical nature of amino acid substitutions, population frequency (ESP and 1000 genomes), and predicted functional impact (including PolyPhen [15] and SIFT [16] in silico prediction tools).
This was followed by stepwise filtering and removal of common SNPs, intergenic and 3'/5' UTR variants, non-splice-related intronic variants, and synonymous variants (except those at the first and last nucleotide position of an exon). All variants annotated within the HGMD and/or the OMIM databases were protected during this filtration process. Family history and inheritance models were then applied for further filtration of these variants. Inheritance models executed for each family included autosomal dominant (AD), autosomal recessive (AR), X-linked recessive (XLR), X-linked dominant (XLD), Y-linked (YL) inheritance in male probands, as well as X-linked and autosomal reduced penetrance models [4]. Sequence alignments of the reads were viewed using the Integrative Genomics Viewer (IGV) [17] software. Details about the interpretation of IGV output images can be found at http://www.broadinstitute.org/software/igv/AlignmentData.
Personalized medical review with enhanced and comprehensive assessment [PRECISE] of potentially causal alterations, variant confirmation and cosegregation analysis
Variants were comprehensively assessed by a molecular geneticist to identify the most likely causative mutation [s] as previously described [4]. These were additionally reviewed by another molecular geneticist and/or a genetic counselor before reporting. Amplification primers were designed using PrimerZ [18] and sequencing was performed on an ABI3730 (Life Technologies, Carlsbad, CA) using standard procedures. Co-segregation Sanger analysis was performed when additional family members were available and segregation data could assist with interpretation.
Results
Sequence alignment and variant calling
In 2,703 previous clinical samples (patients and informative family members) that were run through the Ambry Exome pipeline, a total of 33,944,032 indels were detected that passed the read depth cutoff of 10X, and variant calling quality score (Q-score) of 20 (Table 1). Among these, the overwhelming majority (~94%) was in the size range of 1-10 base pairs (bp), and the percentage of indels decreased drastically as the size of the indel increased beyond 10bp (Table 1). The largest insertion and deletion detected using the Ambry Exome pipeline were 236 bp and 300 bp, respectively.
![]()
Table 1: Numbers of Indels called by the Ambry Exome Pipeline before filtration. % indicates the percentage of total insertions or total deletions detected, respectively.
View Table 1
Variant filtration, FIND, PRECISE, causal variant reporting
Positive/Likely positive findings in characterized as well as novel genes were reported in 284 (28%) of the first 1000 patients referred to Ambry Genetics for DES. Out of these, 114 (40%) were small deletions and insertions with the majority of them in the < 10 bp size range (Table 2). A colocalized indel (i.e. one with both an insertion and a deletion) was included in each category. Deletions were seen to be pathogenic at a higher frequency than insertions and the numbers of these variants decreased as the length of the alteration increased.
![]()
Table 2: Indels as diagnostic mutations reported in the first 1000 patients undergoing DES. % indicates the percentage of total insertions or total deletions reported, respectively.
View Table 2
Detection of pathogenic indels larger than 20 base pairs
8 indels larger than 20 bop (Table 3) were clearly pathogenic in terms of their effect in the patients. All 8 indels were confirmed and the exact breakpoints were mapped by Sanger sequencing before reporting. For example, a maternally inherited heterozygous deletion of 89 bp in the UBE3A gene, c.1254_1324del89 (Table 3, Figure 1), was detected in a pediatric patient with a clinical diagnosis of Angelman syndrome (AS), a neurogenetic disorder characterized by severe intellectual and developmental disabilities, seizures, sleep disturbances and hand flapping. The UBE3A gene encodes the ubiquitin-protein ligase E3A, an enzyme that is involved in ubiquitin-mediated degradation of proteins within cells and loss of this enzyme function has been associated with the characteristic features of AS [19-21]. Both the paternally and maternally inherited copies of this gene are active in all human tissues except the brain where only the maternally inherited copy is expressed [22-24]. Based on this knowledge, it is presumed that the asymptomatic mother's deletion occurred on her paternally inherited allele, which was inactivated due to paternal imprinting.

Figure 1: 89 bp UBE3A deletion (c.1254_1324del89) in proband with Angelman Syndrome inherited from the unaffected mother. The sequence alignments were viewed using the Integrative Genomics Viewer (IGV). For the proband and the mother, these alignments are shown in two panels: the top panel for each shows the ungapped sequence alignment, and the bottom panel shows the presence of a deletion (indicated by a black bar) in approximately half the reads in either direction (colored pink and blue respectively) after the sequences are realigned to the reference sequence using gapped alignment for detection of structural variants. Only one panel is shown for the father since the ungapped sequence showed perfect alignment to the reference sequence indicating an absence of an insertion or deletion in this chromosomal region.
View Figure 1
![]()
Table 3: Reported Pathogenic Indels larger than 20 bp.
View Table 3
In another case, a heterozygous 58 bp deletion in the RFXANK gene (c.419_438+38del58) was detected in a pediatric patient with recurrent infections, immunodeficiency and significantly decreased expression of CD127 (Table 3, Figure 2). Mutations in the RFXANK gene are associated with MHC Class II deficiency of the complementation group B and are usually inherited in an autosomal recessive manner [25]. Initial Exome analysis of the proband detected a homozygous splice site mutation (15/15 mutant reads, Q score 42), c.438+1G > T, in the RFXANK gene. A close look at the IGV data revealed that the c.419_438+38del58 deletion on the other allele encompasses the splice site substitution and thus makes the latter apparently homozygous (Figure 2). Cosegregation analysis confirmed that the proband and similarly affected identical twin inherited one alteration each from the unaffected parents.
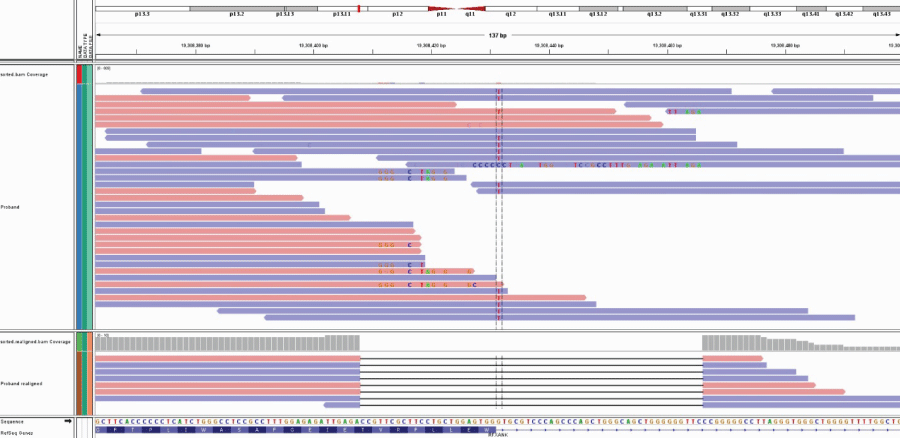
Figure 2: Sequence alignments (ungapped in top panel and gapped in the bottom panel) of the RFXANK compound heterozygous mutations - splice site mutation (c. 438+1G > T bracketed in dashed vertical lines in the ungapped alignment) and 58 bp deletion (c.419_438+38del58 indicated by a black bar in the gapped alignment) in proband with MHC Class II deficiency of the complementation group B. Sequence alignments to the reference sequence were viewed using the Integrative Genomics Viewer (IGV).
View Figure 2
Discussion
Indels have been frequently implicated in human genetic disease, especially those that disrupt the mRNA open reading frame and lead to the nonsense-mediated decay of the mutant mRNA or production of truncated proteins. However, accurate calling of indels larger than 20 base pairs (bp) using NGS based approaches for whole genome and whole exome analysis remains a challenge due to alignment errors, repeat sequences, incomplete reference genome, accuracy issues and unreliability of sequencing and bioinformatics analysis in the clinical diagnostic setting. Over the last few years, a variety of NGS indel calling pipelines such as the GATK Unified Genotyper [26], SOAPindel [27], and SAMtools [28] have been developed. However, a low concordance rate among indels called by these pipelines has been reported with only 26.8% of them being called by all three [29].
Recently, a comparison between an alignment-based indel calling algorithm such as the GATK Unified Genotyper, and a newly developed assembly-based indel calling algorithm called Scalpel [30] demonstrated that a higher depth of sequencing coverage is required to improve the sensitivity of detecting indels that are greater than 5 bp in length [10]. However, the authors found that even at a 90X depth of coverage, only 52% of large indels were called by the GATK Unified Genotyper, whereas Scalpel was successful in calling 90% of large indels. This indicated that although an assembly based approach was more efficient than an alignment based approach in accurately mapping larger indels, higher coverage over the entire length of the indel was also necessary in order to have sufficient numbers of reads for an efficient micro-assembly for indel calling.
A careful analysis of all the variant calling pipelines available when we introduced diagnostic exome sequencing in 2011 led us to incorporate CASAVA 1.8 (Illumina, Inc., San Diego, CA) into the Ambry Exome pipeline. While GATK was the most widely used pipeline for whole exome analysis, a comparison with CASAVA1.8 on chromosome 21 of a Yoruba trio indicated that there was no significant difference between the two pipelines in terms of SNP calling [31]. In contrast, the authors found CASAVA1.8 to be more conservative in indel calling as compared to GATK - approximately 12000 indels were called by CASAVA1.8 (close to the 12,558 INDELS per individual called by the Ambry Exome pipeline - 15950031/2703 exomes), while GATK, that runs the Dindel [32] program for indel detection, called about 16000 indels per individual. However, Dindel is designed for small indels whereas CASAVA1.8 uses an algorithm that combines both gapped alignments, which efficiently identify relatively small indels (1-10 bases), and contig assembly and alignments, which can efficiently identify much larger indels (> 10 bases).
In a recent review of bioinformatics tools by Daber, et al. [33] detection of indels as large as 90bp was reported using tools including Novoalign, GATK and AbsoluteVar [33]. In comparison, using the Ambry Exome pipeline, we were able to detect insertions and deletions as large as 236bp and 300bp, respectively. We believe that our custom bioinformatics workflow, followed by annotation and variant analysis using the Ambry AVA software and phenotype-based manual review is responsible for this enhancement in the detection of longer indels.
In conclusion, indels larger than 20 bp account for 3.2% of the positive cases in our DES cohort. Further, we report what is, to our knowledge, the largest deletion (204 bp, Table 3) detected by DES and confirmed by Sanger sequencing to date. An optimized workflow from sequence alignment and variant calling to family-based genetic model filtering and manual critical review can reliably identify and prioritize deletions up to 200 bp in size. Although detection of indels of such sizes is inherently difficult for DES, this functionality is essential for enhanced clinical sensitivity and improved diagnostic yield.
Acknowledgments
We are grateful to the patients and their families are for their participation.
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
ESP (Internet): Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA (URL: http://evs.gs.washington.edu/EVS/) (accessed May, 2015).
OMIM (Internet): Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University, Baltimore, MD (URL: http://omim.org/) (accessed May, 2015)
PubMed [Internet]: National Center for Biotechnology Information, U.S. National Library of Medicine, Bethesda MD (URL: http://www.ncbi.nlm.nih.gov/pubmed) (accessed May, 2015)
References
-
Rabbani B, Tekin M, Mahdieh N (2014) The promise of whole-exome sequencing in medical genetics. J Hum Genet 59: 5-15.
-
Yang Y, Muzny DM, Xia F, Niu Z, Person R, et al. (2014) Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 312: 1870-1879.
-
Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, et al. (2014) Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 312: 1880-1887.
-
F Farwell KD, Shahmirzadi L, El-Khechen D, Powis Z, Chao EC, et al. (2014) Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med 17: 578-586.
-
Majewski J, Schwartzentruber J, Lalonde E, Montpetit A, Jabado N (2011) What can exome sequencing do for you? J Med Genet 48: 580-589.
-
Dolled-Filhart MP, Lee M Jr, Ou-Yang CW, Haraksingh RR, Lin JC (2013) Computational and bioinformatics frameworks for next-generation whole exome and genome sequencing. ScientificWorldJournal 2013: 730210.
-
Bao R, Huang L, Andrade J, Tan W, Kibbe WA, et al. (2014) Review of current methods, applications, and data management for the bioinformatics analysis of whole exome sequencing. Cancer Inform 13: 67-82.
-
International HapMap 3 Consortium, Altshuler DM, Gibbs RA, Peltonen L, Altshuler DM, et al. (2010) Integrating common and rare genetic variation in diverse human populations. Nature 467: 52-58.
-
Mullaney JM, Mills RE, Pittard WS, Devine SE (2010) Small insertions and deletions (INDELs) in human genomes. Hum Mol Genet 19: R131-136.
-
Fang H, Wu Y, Narzisi G, O'Rawe JA, Barrón LT, et al. (2014) Reducing INDEL calling errors in whole genome and exome sequencing data. Genome Med 6: 89.
-
Karakoc E, Alkan C, O'Roak BJ, Dennis MY, Vives L, et al. (2011) Detection of structural variants and indels within exome data. Nat Methods 9: 176-178.
-
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, et al. (2001) dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29: 308-311.
-
1000 Genomes Project Consortium, Abecasis GR, Altshuler D, Auton A, Brooks LD, et al. (2010) A map of human genome variation from population-scale sequencing. Nature 467: 1061-1073.
-
Stenson PD, Mort M, Ball EV, Howells K, Phillips AD, et al. (2009) The Human Gene Mutation Database: 2008 update. Genome Med 1: 13.
-
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, et al. (2010) A method and server for predicting damaging missense mutations. Nat Methods 7: 248-249.
-
Ng PC, Henikoff S (2006) Predicting the effects of amino acid substitutions on protein function. Annu Rev Genomics Hum Genet 7: 61-80.
-
Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, et al. (2011) Integrative genomics viewer. Nat Biotechnol 29: 24-26.
-
Tsai MF, Lin YJ, Cheng YC, Lee KH, Huang CC, et al. (2007) PrimerZ: streamlined primer design for promoters, exons and human SNPs. Nucleic Acids Res 35: 63-65.
-
Kishino T, Lalande M, Wagstaff J (1997) UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet 15: 70-73.
-
Matsuura T, Sutcliffe JS, Fang P, Galjaard RJ, Jiang YH, et al. (1997) De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat Genet 15: 74-77.
-
Lossie AC, Whitney MM, Amidon D, Dong HJ, Chen P, et al. (2001) Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J Med Genet 38: 834-845.
-
Albrecht U, Sutcliffe JS, Cattanach BM, Beechey CV, Armstrong D, et al. (1997) Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat Genet 17: 75-78.
-
Rougeulle C, Glatt H, Lalande M (1997) The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain. Nat Genet 17: 14-15.
-
Vu TH, Hoffman AR (1997) Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat Genet 17: 12-13.
-
Masternak K, Barras E, Zufferey M, Conrad B, Corthals G, et al. (1998) A gene encoding a novel RFX-associated transactivator is mutated in the majority of MHC class II deficiency patients. Nat Genet 20: 273-277.
-
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, et al. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491-498.
-
Li S, Li R, Li H, Lu J, Li Y, et al. (2013) SOAPindel: efficient identification of indels from short paired reads. Genome Res 23: 195-200.
-
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078-2079.
-
O'Rawe J, Jiang T, Sun G, Wu Y, Wang W, et al. (2013) Low concordance of multiple variant-calling pipelines: practical implications for exome and genome sequencing. Genome medicine 5: 28.
-
Narzisi G, O'Rawe JA, Iossifov I, Fang H, Lee YH, et al. (2014) Accurate de novo and transmitted indel detection in exome-capture data using microassembly. Nat Methods 11: 1033-1036.
-
Bauer DC (2011) Variant calling comparison CASAVA1.8 and GATK. Nature Precedings.
-
Albers CA, Lunter G, MacArthur DG, McVean G, Ouwehand WH, et al. (2011) Dindel: accurate indel calls from short-read data. Genome Res 21: 961-973.
-
Daber R, Sukhadia S, Morrissette JJ (2013) Understanding the limitations of next generation sequencing informatics, an approach to clinical pipeline validation using artificial data sets. Cancer Genet 206: 441-448.
