

Journal of Genetics and Genome Research
Development of a Comprehensive NGS Workflow for the Analysis of Tumor BRCA1 and BRCA2 Mutations and Large Rearrangements
Zhengwei Dong1#, Hua Dong1#, Xiaorong Zhong2, Zuxiang Peng2, Xuehua Zhu1, Yun Sun1, Yunqin Chen3, Changting Liu1, Xiaolu Yin1, Guanshan Zhu1, Hong Zheng2 and Yi Gu1*
1Asia and Emerging Markets iMed, AstraZeneca, Zhangjiang Hi-Tech Park, Shanghai 201203, P. R. China
2Laboratory of Molecular Diagnosis of Cancer, Cancer Center, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, P. R. China
3R&D Informatics, AstraZeneca, Zhangjiang Hi-Tech Park, Shanghai 201203, P. R. China
#Equal contributors
*Corresponding author:
Yi Gu, Asia and Emerging Markets iMed, AstraZeneca, Zhangjiang Hi-Tech Park, Shanghai 201203, P. R. China, Tel: +86 21 61097850, Email: Yi.Gu@astrazeneca.com
J Genet Genome Res, JGGR-2-019, (Volume 2, Issue 2), Research Article; ISSN: 2378-3648
Received: June 29, 2015 | Accepted: September 24, 2015 | Published: September 28, 2015
Citation: Dong Z, Dong H, Zhong X, Peng Z, Zhu X, et al. (2015) Development of a Comprehensive NGS Workflow for the Analysis of Tumor BRCA1 and BRCA2 Mutations and Large Rearrangements. J Genet Genome Res 2:019. 10.23937/2378-3648/1410019
Copyright: © 2015 Gu Y, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Patients with germline or somatic BRCA1 and BRCA2 mutations are sensitive to PARP inhibitor treatment. However, current clinical testing of BRCA1/2 is limited to germline mutations in blood samples. In the present study, we have developed and validated a workflow for BRCA1/2 mutation test in patient tumor samples, which can identify both germline and somatic mutations. Our approach combined targeted capturing with the BRCA MASTR assay and consequent sequencing using Miseq, a benchtop next-generation sequencing (NGS) platform. The cut-offs for calling substitution mutations and large rearrangements were defined using 45 normal human samples, and then evaluated using 9 human cell lines harboring known BRCA1/2 mutations, as well as 100 patient tumor tissue samples. At a sensitive mutant allele frequency threshold of 5%, all somatic mutation calls were confirmed using an independent technology. Using amplicon-specific cut-offs for large deletion calls, one out of two large deletion candidates was confirmed by the long-range PCR. In summary, our workflow offers a useful tool for testing tumor BRCA1/2 mutations including substitutions, small insertions or deletions (indels) and large deletions.
Keywords
NGS, BRCA1, BRCA2, Mutation, Large rearrangement
Background
The BRCA1 and BRCA2 genes are two key components of the homologous recombination repair (HRR) pathway in DNA double-strand breaks (DSBs) repair [1]. Deleterious mutations in the BRCA1/2 genes lead to an increased risk for breast or ovarian cancer as part of hereditary breast-ovarian cancer syndrome [2]. In many countries, BRCA1/2 gene testing of blood samples has become routine practice for patients with family history of breast and ovarian cancer. Decision-making for prevention or treatment of these tumor types heavily rely on gene test results. Recently, preclinical and clinical studies have shown that the BRCA1/2 mutation carriers are more sensitive to poly adenosine diphosphate (ADP) ribose polymerase (PARP) inhibitors such as Olaparib and Veliparib [3-7]. Clinical benefit of PARP inhibitors has also been validated in ovarian cancer patients with germline and somatic BRCA1/2 mutations [5]. Therefore, BRCA1/2 could serve as a critical biomarker for patient selection in PARP inhibitor treatment. However, somatic mutations in tumors are not detected by blood testing, which is currently the mainstream method. Direct testing in tumor samples is in great need to identify additional patients with somatic mutations that may also benefit from PARP inhibitor treatment.
As BRCA1 and BRCA2 are very long genes lacking mutation hotspots, direct sequencing of these genes are of great interest. On the technology side, next-generation sequencing (NGS) technologies, based on parallel sequencing of individual DNA fragments, are setting the new standard in speed and cost effectiveness of genome sequence analysis. Benchtop NGS platforms like the Illumina MiSeq are feasible options for routine clinic use with short turnaround time and high cost effectiveness. Current bottleneck for NGS gene testing is in data analysis. Although several mutation calling pipelines have recently been developed for the BRCA1/2 gene testing by NGS method [8-18], they mostly focused on detection of germline BRCA1/2 mutations from blood samples. So far, only two pipelines were reported for somatic mutation detection from tumor samples. Frampton et al. [18] developed and validated an NGS panel of 287 cancer related genes from formalin-fixed and paraffin-embedded (FFPE) tumor tissue samples [18]. Pritchard et al. [8] developed the UW-OncoPlex assay for comprehensive analysis of 194 clinically relevant cancer genes [8]. However, these two NGS workflow and mutation calling pipelines were not designed and validated for the extremely complicated patterns of genomic variants in BRCA1/2 genes. To enhance patient selection for PARP inhibitor therapy, there is an emerging need to develop a comprehensive NGS workflow focusing on tumor BRCA1/2 mutation testing.
In the present study, we have selected the BRCA MASTR assay (Multiplicom) coupled with the benchtop MiSeq platform. In additional to the experimental workflow, we have also developed comprehensive bioinformatics pipelines for calling BRCA1/2 mutations including substitutions, small insertions or deletions (indels) and large rearrangements from freshly frozen human normal tissues. This workflow was then validated using human cell lines with the known BRCA1/2 mutations, and eventually tested on a cohort of tumor tissue samples from 100 breast cancer patients.
Methods
Patients and samples
Frozen tissues adjacent to tumors from 45 patients were generously provided by Zhongshan hospital and Beijing tumor hospital. These tissues were previously known to have two copies of BRCA1/2 genes by array CGH analysis (data not shown), and therefore were used in this study as normal control to define biological baseline of the workflow. Nine human cell lines from American Type Culture Collection (ATCC) with known BRCA1/2 mutations and three commercially available normal human DNA samples (Promega, USA) were used to validate the workflow. Furthermore, 100 patients who were diagnosed with breast cancer from Feb. 2008 to Sep. 2013 at West China Hospital were recruited into this study for BRCA1/2 mutation testing and validation. All fresh frozen tissues from breast cancer patients were reviewed by the pathologist to ensure a tumor content of more than 50%. The human study was approved by the Ethics Committee of the hospitals, and the written informed consent was provided by the patients.
DNA extraction and quality check
DNA was extracted using Gentra Puregene kit (QIAGEN, Germany) following the manufacturer's instructions. DNA concentration and purity were assessed using Nanodrop 2000 (Thermo Fischer Scientific, USA), and all DNA samples had A260/280 nm ratio between 1.8-2.0.
Multiplex PCR based targeted sequencing
The coding exons and flanking regions of BRCA1 and BRCA2 genes were enriched using BRCA MASTR assay v2.1 (Multiplicom, Belgium), which contains 93 primer pairs in five reactions, according to the manufacturer's instructions. Briefly, a target-specific multiplex PCR was performed, followed by a second PCR with universal primers to integrate sequencing adaptors and indexing tags. The universal PCR amplified products were purified with the Agencourt AMPure XP kit (Beckman Coulter Genomics, USA), quantified with the Quant-iT dsDNA BR Assay kit using Qubit® 2.0 Fluorometer (Life Technologies, USA), pooled at equal nanomolar and sequenced using MiSeq Reagents v2.0 (2 × 250 bp pair-end sequencing) on MiSeq system (Illumina, USA).
Bioinformatics analysis
The informatics pipeline was shown in figure 1. The region of interest (ROI) was defined as the coding exon sequence plus 20 bp in the preceding intron and 10 bp in the following intron to ensure correct analysis of consensus splicing sites. Primer sequences were trimmed off from raw reads using custom scripts. Mapping, substitution mutation and indel calling are completed in Array Studio v7.2 (OmicSoft, USA), a commercially available software. The raw reads were aligned to reference human genome GRCh37.3, and non-uniquely mapped reads were then removed. The size detection window for small indels was set up to 60 bp, and the minimum amplicon coverage was set to 500×. All variants were called using reference transcripts NM_007294.3 for BRCA1 and NM_000059.3 for BRCA2, respectively. Base quality was set to Q30, and mutant allele frequency cut-off setting was described in details in the Results section. The variants were then filtered by dbSNP138 to remove common SNPs, and all variant nomenclature follows the Human Genome Variation Society (HGVS) guidelines.
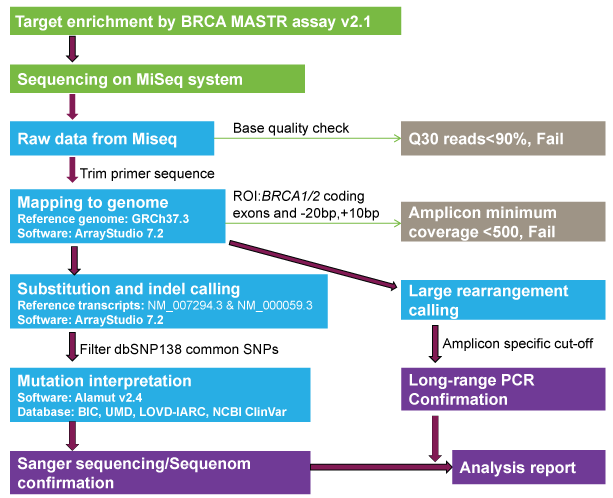
Figure 1: NGS workflow for tumor BRCA1/2 mutation testing
Experimental process is shown in the green box. Quality control process is shown in gray box. Informatics analysis process is shown in blue box. Wet lab confirmation and analysis report are shown in purple box.
View Figure 1
Customized scripts were developed to call large rearrangements in BRCA1/2 genes as shown in the Results section.
Confirmation of substitution mutations and indels
All substitution mutation and indel candidates with mutant allele frequency > 0.2 were confirmed by Sanger sequencing. Briefly, each candidate was amplified using MASTR assay primer pair that covers the specific mutation site. The PCR products were sequenced in both directions on an ABI 3730XL DNA analyzer (Life Technologies, USA).
Candidates with allele frequencies between 0.05-0.2 were confirmed by the Mass ARRAY system (Sequenom, USA). Briefly, genomic DNA amplification and single base pair extension were conducted using specific primers that were designed by Sequenom Assay Designer v3.1 software. The allele-specific single base extension products were then quantitatively analyzed using matrix-assisted laser desorption/ionization-time of flight/mass spectrometry (MALDI-TOF/MS) on the Sequenom Mass Array Spectrometer.
Large rearrangement confirmation
The confirmation of large rearrangement candidates from NGS analysis were performed by long-range PCR, followed by Sanger sequencing and primer walking. Specific primers were designed to span the target exons and flanking introns (Supplementary Table 1). The templates were amplified using LA PCR Kit (TaKaRa, Japan). The PCR products were sequenced by a step-by-step primer walking approach to sequence the long DNA templates from end to end on an ABI 3730XL DNA analyzer.
Mutations interpretation
All confirmed variants were interpreted following guidelines from both the American College of Medical Genetics and Genomics (ACMG) and International Agency for Research on Cancer (IARC) guidelines [19,20]. We collected the evidences for interpretation by the Alamut software (Interactive biosoftware, France) in combination with databases including BIC (http://research.nhgri.nih.gov/bic/), UMD (http://www.umd.be/), NCBI ClinVar database (http://www.ncbi.nlm.nih.gov/clinvar/) and LOVD-IARC (http://brca.iarc.fr/LOVD/home.php). All variants were classified into 5 classes, and the pathogenic and likely pathogenic mutations were considered as deleterious mutations.
Results
Experimental design
This study aims to establish a comprehensive NGS workflow for analyzing tumor BRCA1/2 substitution mutation, indel and large rearrangement as shown in figure 1. For the targeted sequencing of BRCA1/2 genes, we have selected the BRCA MASTR assay coupled with an Illumina MiSeq sequencing platform, after evaluating various assays and sequencing platforms.
One main goal of this workflow is to enable the calling of somatic mutations with low allele frequencies in human tumor samples using NGS. In addition, we aim to develop a large rearrangement calling pipeline using NGS data. We used the BRCA1/2 gene sequencing data from 45 human normal tissues to define the cut-offs for mutation and large rearrangement calling. Then, we validated our workflow using 9 human cell lines with known BRCA1/2 mutations and 100 primary human tumor samples.
Substitution mutation and small indel calling pipeline
To define the mutant allele frequency cut-off, all nucleotides different from the reference allele on human genome GRCh37.3 were called from the 45 human normal tissues. Common polymorphisms were then filtered out against the dbSNP138 database. Rare variants which are in the dbSNP138 database were also removed. The remaining variants were considered as sequencing errors. The highest error rates of each site in the BRCA1/2 region of interest (ROI) among 45 samples were shown in figure 2. We found that sequencing error rates were locus-specific and nucleotide-specific. A-G and T-C changes have the highest error rate, while the G-C and A-T changes were the lowest. Across about 24K target regions, the error rates for each position vary from 0.01% to 3.9%. Considering the sensitivity limit of currently available confirmation technologies such as Sequenom mass array used in this study, we set the allele frequency cut-off at 5% for substitution mutation calling in the whole ROI.
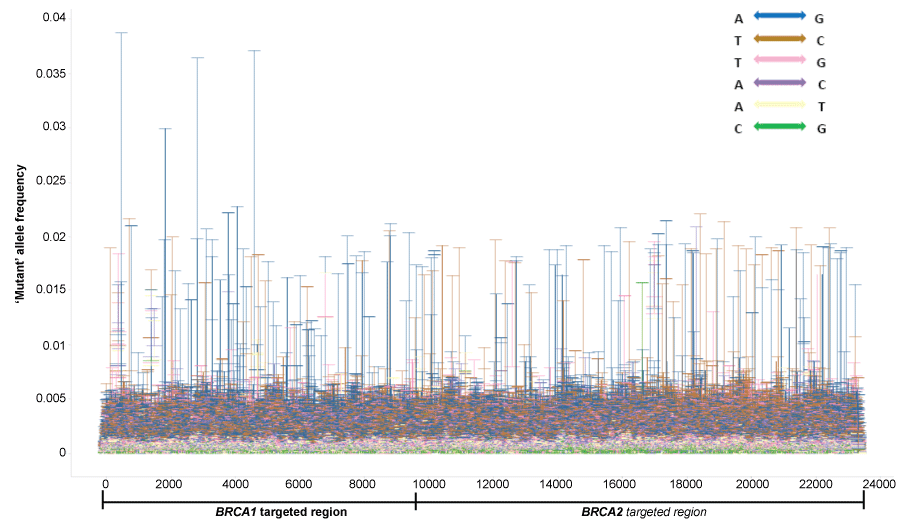
Figure 2: Distribution of locus-specific 'mutant' allele frequency in normal samples
'Mutant' allele frequency distribution in 45 human normal samples were plotted across the BRCA1/2 targeted region (about 24 Kb) after removal of common SNPs. Maximum allele frequencies, which reflects the error rate of each site, were shown as bars, and each nucleotide type change was shown in a different color.
View Figure 2
For small indel calling, the background error rate was higher than the substitution mutation. For example, the long homopolymers in BRCA1/2 coding regions and intron-exon boundaries led to a false 1 bp deletion. Therefore, we manually visualized the variants in Array Studio genome browser to remove some such sequencing artifacts and software mapping artifacts. For the remaining indel variants, we also set the allele frequency cut-off at 5% and then confirmed them by Sanger Sequencing or Mass ARRAY system.
Large rearrangement calling pipeline
For BRCA1/2 gene large rearrangement calling, we used 45 human normal tissue samples to determine the normal range of copy number variation of BRCA1/2 gene exons. These 45 samples were previously known to have two copies of the BRCA1/2 genes by array CGH analysis (data not shown). We calculated the ratio of each of the 93 amplicons in each sample based on normalized amplicon coverage.
For sample i, Amplicon j, Sample size N
For each amplicon, the mean value and range of the ratios in normal samples were summarized in figure 3. The mean values for all amplicons are close to 1, consistent with previous array CGH data. However, the range of ratios varies across amplicons, and the distributions were not always normal. Therefore, we set amplicon specific cut-offs for the ratios by random sampling and permutation. For each amplicon, the 99% confidence interval was taken as the minimum and maximum of acceptable amplicon ratios.
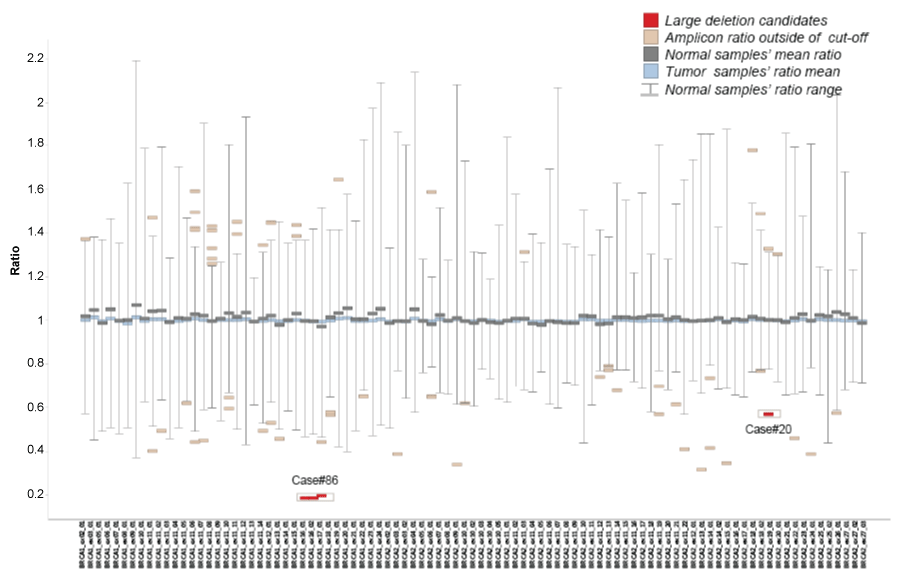
Figure 3: Amplicon-specific ratio distribution in 100 tumor samples
Amplicon ratios were calculated from 45 human normal samples and 100 tumor samples. Mean, minimum and maximum ratios for each amplicon in normal samples were shown as the gray bars. Mean amplicon ratio in 100 tumor samples were shown as the cyan bars. Amplicon ratios outside of the normal range but filtered against the two exclusion criteria were shown as the light brown bars. The remaining candidates were shown as the red bars.
View Figure 3
In addition, as reported large rearrangements in BRCA1/2 genes primarily consist of the deletions of one or more consecutive exons [21], we have also excluded: 1) Samples with multiple inconsecutive amplicons with ratios beyond cut-offs. 2) Samples with a single amplicon beyond cut-offs, covering only part of one exon. The remaining candidates were selected for long-range PCR confirmation.
Cell line validation
Due to the absence of patient tumor samples with known BRCA1/2 mutations, we used nine cell lines with known BRCA1/2 mutations to validate the BRCA1/2 mutation testing workflow described above, including targeted sequencing and mutation calling. Three commercially available normal human DNA samples were used as the negative control. As shown in table 1, our workflow detected all known substitution mutations and indels accurately in these cell lines. Because there are no commercial available cell lines with known BRCA1/2 large rearrangements, we did not validate the large rearrangement calling pipeline through this approach.
![]()
Table 1: BRCA1/2 mutations from nine human cell lines.
View Table 1
Tumor sample testing
Next we tested our workflow with tumor samples from 100 breast cancer patients. Using 5% mutant allele frequency cut-off, we called a total of 33 BRCA1/2 substitution mutations and indels (Table 2). Among them, 8 mutations are classified as pathogenic. All of the 33 mutations were fully confirmed by a second platform such as Sanger sequencing or Sequenom mass array (Supplementary Figure 1).
![]()
Table 2: BRCA1/2 mutations from 100 breast cancer patients.
View Table 2
For large rearrangement calling, the amplicon ratios from these 100 tumor samples were shown in figure 3. In total, 64 amplicons in 17 samples were found with ratios outside of the pre-defined normal ranges. After further filtering against the two exclusion criteria set above, only 4 amplicons in 2 samples were called. The 2 candidates include Case #86 with a deletion of 3 consecutive amplicons deletion and Case #20 with a single amplicon deletion. Among these two, only the deletion of 3 consecutive amplicons over exons 16 and 17 of (c.4676_5074del) BRCA1 (Figure 4A and Figure 4B) in case #86 was confirmed by the long range PCR (Figure 4C). The breakpoint junction sequences of this large deletion were also shown in figure 4D. BRCA1 exon 16-17 deletion has been reported [21,22] and classified as pathogenic in the UMD database [23], and our long range PCR further defined the exact breakpoints of the deletion, which were never reported before.
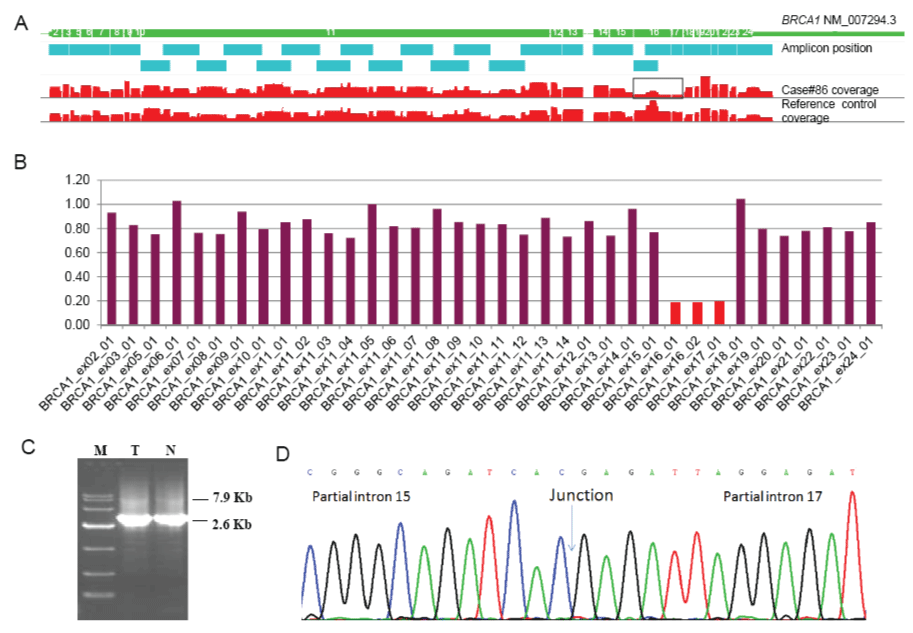
Figure 4: BRCA1 exon16-17 deletion (c.4676_5074del) calling and confirmation in the Case #86
(A) BRCA1 exon coverage plot. Case #86 showed much less coverage in the exons 16 and 17 than reference control sample.
(B) Amplicon ratios of Case #86. Three amplicons covering the BRCA1 exons 16 and 17 (NM_007294.3) showed ratios close to 0.2.
(C) Gel image of long-range PCR products. The expected band size of normal allele is 7.9 Kb. The band size of allele with exon16-17 deletion is 2.6 Kb. M, marker DL10000; T, tumor DNA sample; N, paired normal sample.
(D) Sanger sequencing image of the breakpoint flanking sequences with exons 16 and 17 deletion. The sequence before the breakpoint is on BRCA1 intron 15, and the sequence after the breakpoint is on BRCA1 intron 17.
View Figure 4
Discussion
In current clinical practice, BRCA1 and BRCA2 testing has been mainly focused on germline mutations in blood samples. The mutant allele frequency cut-off at 15%-20% is sufficient to capture all heterozygous germline mutations [13-15]. This is relatively easier as compared to detection of low frequency somatic mutations in tumor tissues due to tumor heterogeneity. Recent studies showed that patients with somatic BRCA1/2 mutations are equivalently sensitive to PARP inhibitor treatment as patients with germline BRCA1/2 mutations [5]. Thus, BRCA1/2 mutation testing with tumor samples, including both germline and somatic mutations, could help more patients who would benefit from PARP inhibitor therapy. In this study, we developed a comprehensive NGS workflow for detection and analysis of tumor BRCA1/2 mutations including substitutions, indels and large deletions, using a multiplex PCR technology coupled with a benchtop NGS platform and a customized bioinformatics pipeline. Our results indicate that this workflow achieves a good performance for tumor BRCA1/2 mutation testing at 5% mutant allele frequency cut-off.
For tumor BRCA1/2 mutation screening, the first essential step is to select an efficient assay and a sequencing platform. We have evaluated both Miseq and PGM platforms, as well as three different assays including BRCA MASTR assay, AmpliSeq BRCA panel and a customized Haloplex BRCA panel (data not shown). We found that MASTR BRCA assay coupled with MiSeq sequencing platform had the best performance in terms of data quality and cost effectiveness. We have also compared different variant callers including GATK [24], Varscan [25], Freebayes [26] and Array Studio for the deep coverage of BRCA1/2 targeted sequencing datasets. We got identical calling results except for minor differences on mutant allele frequencies and nomenclature (data not shown). We chose Array Studio pipeline for its accuracy in mapping and variant calling, and user-friendly interface.
After a systematic evaluation of error rates of all amplicons, we set the cut-off of mutant allele frequency at >= 5% for substitution mutation and indel calling, which had 100% confirmation rate, suggesting superior specificity. The pipeline developed by Pritchard et al. [8] focusing on 194 clinically relevant genes including BRCA1/2 had 99.2% sensitivity on mutant allele frequency >= 10% and 67% sensitivity on mutant allele frequency < 10% [8]. Frampton et al. [18] also designed a panel of 287 cancer related genes including BRCA1/2 and showed 100% sensitivity on mutant allele frequency >= 10% and 99% sensitivity on mutant allele frequency < 10% [18]. Our workflow demonstrated the capability to detect BRCA1/2 mutations in frozen tumor sample with mutant allele frequency >= 5%. For mutations with allele frequencies < 5%, further studies are warranted for validation using more sensitive confirmation technologies.
A minority of BRCA1/2 mutations are large rearrangements of DNA segments that disrupt gene function [21]. Traditionally, Multiplex ligation-dependent probe amplification (MLPA) or qPCR assay was used to detect exonic copy number changes in blood, saliva or FFPE samples. More recent studies use NGS workflow to detect gene copy number variations (CNVs) or large rearrangements [27,28]. Usually, a unified cut-off was set for all amplicons, for example, < 0.6 for deletions and > 1.4 for duplications [10,14,15,29]. However, for multiplex PCR methods, due to different PCR amplification efficiency of each primer pair, the amplicon coverage and their corresponding variance are different. Therefore, we defined amplicon specific cut-offs based on amplicon specific ratio ranges derived from human normal samples. In this study, the candidate with a deletion of three consecutive amplicon (ratio around 0.2) was confirmed by long rang PCR, while the candidate with a single amplicon 'deletion' (ratio around 0.5) was not. Taken together, our NGS workflow could serve as a screening method for large rearrangement calling from tumor tissues, which may need further confirmation by a second technology like long-range PCR or MLPA.
In terms of assay robustness, our workflow could load maximum 48 samples in a single run, and complete the experiment process, informatics analysis and mutation interpretation within two weeks. This meets most of BRCA1/2 mutation testing needs in clinical practice.
Our NGS workflow here offers a useful tool for testing germline and somatic BRCA1/2 mutations including substitutions, small indels and large deletions in freshly frozen tumor tissues. It combines a benchtop MiSeq NGS platform and improved calling sensitivity. We have developed a bioinformatics pipeline customized for the BRCA1 and BRCA2 genes from QC to mapping, substitution/ indel calling and large rearrangement detection. In addition, the workflow has the potential to be applied to other tumor suppressor genes like TP53 that does not have mutation hotspots.
Competing Interests
Zhengwei Dong, Hua Dong, Xuehua Zhu, Yun Sun, Yunqin Chen, Changting Liu, Xiaolu Yin and Yi Gu are employees of AstraZeneca during this study. No other conflicts of interests to declare.
Authors' Contributions
ZD and HD were responsible for generating and analyzing the data, and drafting the manuscript; XRZ, ZP and HZ for patient sample collection; XHZ, YS and YC for related data confirmation and interpretation. CL and XY for pathology review of the patient tissues, GZ for study design and technical discussion, and YG for overall study design, drafting and finalizing the manuscript. All authors have read and approved the final version.
Acknowledgements
We thank Drs. Min Hu, Haiyan Xu and Meizhuo Zhang from AstraZeneca for reviewing this manuscript. We are grateful to Dr. Andrew Wallace from Saint Mary's Hospital (Manchester) for the helpful instructions for BRCA1/2 mutation interpretation.
References
-
Narod SA, Foulkes WD (2004) BRCA1 and BRCA2: 1994 and beyond. Nat Rev Cancer 4: 665-676.
-
King MC, Marks JH, Mandell JB; New York Breast Cancer Study Group (2003) Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 302: 643-646.
-
Lee JM, Ledermann JA, Kohn EC (2014) PARP Inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann Oncol 25: 32-40.
-
Plummer R (2010) Perspective on the pipeline of drugs being developed with modulation of DNA damage as a target. Clin Cancer Res 16: 4527-4531.
-
Pennington KP, Walsh T, Harrell MI, Lee MK, Pennil CC, et al. (2014) Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res 20: 764-775.
-
Alsop K, Fereday S, Meldrum C, deFazio A, Emmanuel C, et al. (2012) BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. J Clin Oncol 30: 2654-2663.
-
Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, et al. (2014) Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol 15: 852-861.
-
Pritchard CC, Salipante SJ, Koehler K, Smith C, Scroggins S, et al. (2014) Validation and implementation of targeted capture and sequencing for the detection of actionable mutation, copy number variation, and gene rearrangement in clinical cancer specimens. J Mol Diagn 16: 56-67.
-
Hernan I, Borras E, de Sousa Dias M, Gamundi MJ, Mane B, et al. (2012) Detection of genomic variations in BRCA1 and BRCA2 genes by long-range PCR and next-generation sequencing. J Mol Diagn 14: 286-293.
-
Walsh T, Lee MK, Casadei S, Thornton AM, Stray SM, et al. (2010) Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc Natl Acad Sci USA 107: 12629-12633.
-
Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, et al. (2011) Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci USA 108: 18032-18037.
-
Ozcelik H, Shi X, Chang MC, Tram E, Vlasschaert M, et al. (2012) Long-range PCR and next-generation sequencing of BRCA1 and BRCA2 in breast cancer. J Mol Diagn 14: 467-475.
-
Chan M, Ji SM, Yeo ZX, Gan L, Yap E, et al. (2012) Development of a next-generation sequencing method for BRCA mutation screening: a comparison between a high-throughput and a benchtop platform. J Mol Diagn 14: 602-612.
-
Feliubadalo L, Lopez-Doriga A, Castellsague E, del Valle J, Menendez M, et al. (2013) Next-generation sequencing meets genetic diagnostics: development of a comprehensive workflow for the analysis of BRCA1 and BRCA2 genes. Eur J Hum Genet 21: 864-870.
-
Tarabeux J, Zeitouni B, Moncoutier V, Tenreiro H, Abidallah K, et al. (2014) Streamlined ion torrent PGM-based diagnostics: BRCA1 and BRCA2 genes as a model. Eur J Hum Genet 22: 535-541.
-
Tung N, Battelli C, Allen B, Kaldate R, Bhatnagar S, et al. (2015) Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer 121: 25-33.
-
Castera L, Krieger S, Rousselin A, Legros A, Baumann JJ, et al. (2014) Next-generation sequencing for the diagnosis of hereditary breast and ovarian cancer using genomic capture targeting multiple candidate genes. Eur J Hum Genet 22: 1305-1313.
-
Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, et al. (2013) Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 31: 1023-1031.
-
Plon SE, Eccles DM, Easton D, Foulkes WD, Genuardi M, et al. (2008) Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat 29: 1282-1291.
-
Richards S, Aziz N, Bale S, Bick D, Das S, et al. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405-424.
-
Judkins T, Rosenthal E, Arnell C, Burbidge LA, Geary W, et al. (2012) Clinical significance of large rearrangements in BRCA1 and BRCA2. Cancer 118: 5210-5216.
-
Carvalho M, Pino MA, Karchin R, Beddor J, Godinho-Netto M, et al. (2009) Analysis of a set of missense, frameshift, and in-frame deletion variants of BRCA1. Mutat Res 660: 1-11.
-
Caputo S, Benboudjema L, Sinilnikova O, Rouleau E, Beroud C, et al. (2012) Description and analysis of genetic variants in French hereditary breast and ovarian cancer families recorded in the UMD-BRCA1/BRCA2 databases. Nucleic Acids Res 40: 992-1002.
-
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297-1303.
-
Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, et al. (2012) VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22: 568-576.
-
Garrison E, Marth G (2012) Haplotype-based variant detection from short-read sequencing.
-
Wang C, Evans JM, Bhagwate AV, Prodduturi N, Sarangi V, et al. (2014) PatternCNV: a versatile tool for detecting copy number changes from exome sequencing data. Bioinformatics 30: 2678-2680.
-
Boeva V, Popova T, Lienard M, Toffoli S, Kamal M, et al. (2014) Multi-factor data normalization enables the detection of copy number aberrations in amplicon sequencing data. Bioinformatics 30: 3443-3450.
-
Nord AS, Lee M, King MC, Walsh T (2011) Accurate and exact CNV identification from targeted high-throughput sequence data. BMC Genomics 12: 184.
