

Journal of Genetics and Genome Research
A Perspective on the Algorithms Predicting and Evaluating the RNA Secondary Structure
Giulia Fiscon1*, Giulio Iannello2 and Paola Paci1
1Institute for Systems Analysis and Computer Science "Antonio Ruberti" (IASI), CNR, Italy
2Department of Engineering, University Campus Bio-Medico of Rome, Italy
*Corresponding author:
Giulia Fiscon, Institute for System Analysis and Computer Science "Antonio Ruberti" (IASI), CNR, Via dei Taurini 19, 00185 Rome, Italy, Tel: (+39) 06 49937145, fax (+39) 06 49937106, E-mail: giulia.fiscon@iasi.cnr.it
J Genet Genome Res, JGGR-3-023, (Volume 3, Issue 1), Review Article; ISSN: 2378-3648
Received: February 01, 2016 | Accepted: May 18, 2016 | Published: May 20, 2016
Citation: Fiscon G, Iannello G, Paci P (2016) A Perspective on the Algorithms Predicting and Evaluating the RNA Secondary Structure. J Genet Genome Res 3:023. 10.23937/2378-3648/1410023
Copyright: © 2016 Fiscon G, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Investigating the RNA structure contributes greatly to understand RNA roles in cellular processes. Indeed, functional RNAs show specific instrumental sub-structures for their interaction with other molecules. The RNA structure prediction will provide fundamental insights into developing hypothesis connecting function to structure, but it is a challenging and unsolved task yet.
We aim at discussing the current status of the widespread RNA folding tools and comparing their performances on RNA families with known structure, in order to estimate how much the predictions are close to the experimental folding.
A comprehensive understanding of RNA folding could highlight further roles of long non-coding RNA in the gene expression regulation and in the epigenetic regulatory pathways in physiological and pathological conditions of a living cell.
Keywords
RNA secondary structures, Long non-coding RNA secondary structures, RNA structure prediction tools, Dynamic programming algorithms.
Highlights
• Studying RNA secondary structures represents a cutting-edge topic in structural biology
• Secondary structure predictions are becoming important to connect function to structure
• Understanding of structures may unveil new functions of both coding and non-coding RNAs
• Overview of the existing tools for RNA (coding and non-coding) structure predictions
Introduction
The centrality of RNA molecules in cellular functions has become increasingly evident in recent decades [1,2]. Once regarded only as carriers of genetics information, it has been shown that RNA molecules are functional and play an active role in living organisms: catalysts of metabolic reactions and RNA splicing, regulators of gene expression and guide for protein localization. The linear RNA sequences fold in complex secondary structures (interactions of base-pairs set) whose analysis could be an important determinant of a functional characterization. Indeed, a stable spatial structure of an RNA molecule appears crucial in its interactions with other molecules in the cell [3] and hence, for performing its biological function [4,5]. Thanks to their secondary structure, for instance, the new appreciated long non-coding RNAs (lncRNAs) [6] can guide transcription machinery proteins to specific genomic sites leading to a chromatin remodeling that could allow access of condensed genomic DNA and thus control gene expression. As a consequence, a dysregulation of specific long non-coding RNAs could have a deep impact on cancer development and progression [7].
However, for most RNA sequences the experimental determination of the structure is still arduous. Therefore, the RNA structure prediction is highly requested as well as remains a challenging computational task not wholly solved, yet.
Many tools have been developed to address the prediction of RNA secondary structure based on different methods [8]. Specifically, RNA secondary structures can be determined by using two main approaches: single-sequence [9] and comparative methods [10]. The first class of methods performs prediction starting from single sequences by using techniques that include Free-Energy Minimization (MFE) (e.g., Mfold [11] and RNAfold [12]) and machine learning [13] (e.g., ContextFold [14]); while the second one enables the predictions for sequence families, for example by inferring sets of base-pairs from multiple sequence alignments, looking at the co-variation of nucleotides at different positions.
Among the single-sequence approaches, we focus on the widespread thermodynamic methods, where the stability of a structure is quantified by changes in the folding free energy values according to the nearest neighbor rules [9]. The thermodynamic formula that guides the folding of an RNA molecule is defined as follows:
Where the ratio of the concentration of folded species at equilibrium (F) and the unfolded ones (U) represent the equilibrium constant K. Moreover, ΔG° is the difference between F and U standard free energies [J]; R is the gas constant [J/mol•K] and T the temperature [K]. For equilibrium folding, the lowest free energy structure in the folding ensemble is the most probable [9]. Hence, the aim of predicting secondary structure from thermodynamics is to find the set of base-pairs that provides the lowest free energy reaching the folded state. Alternatively, structures can be sampled from the Boltzmann ensemble according to their probability of occurring, and then can be clustered and the representative structure (called centroid) is determined (e.g., S fold [15]). In addition, other alternative prediction methods rely on the Maximum Expected Accuracy (MEA) structure [16] (i.e., the predicted structure with the highest sum of base paring probability).
Thermodynamic methods can be divided in two main classes: the global folding software (e.g., Fold of RNA structure [17,18], RNA fold of Vienna RNA package [12], Web-Beagle [19] based on a new alphabet to encode secondary structure [20]) and those favoring local folding (e.g., RNALfold [21]). The latter take into account a restriction on the span of base-pairs of the RNA molecule, rather than the structure of the entire RNA and seem to be more accurate since a short-range pairs in long sequences (local folding) are more kinetically favored than long-range pairs (global folding) [22]. It has been shown that thermodynamic models lead to very fast algorithms and reach a high accuracy even if they suffer from steep decrease of accuracy with the increase of sequence length [18]. The drop can be controlled including additional features, such as a partition function (i.e., the sum of the equilibrium constants for all possible secondary structures of a given sequence) to determine the base-pair probabilities of the prediction [23] or searching for homologous sequences to determine a conserved structure [17,24].
A comparative approach to secondary structure prediction exploits multiple sequence alignments to predict a consensus structure shared by all (or most) sequences in the alignment. In particular, given a set of multiple sequences characterized by high sequence conservation, three directions can be followed to predict the lowest free energy structure shared by all the sequences: either (i) firstly a sequence alignment is performed and the information it conveys is then exploited for structure prediction looking at the conserved base-pairs in the found alignment (e.g., RNAalifold [12]); or (ii) the optimal sequence alignment can be found simultaneously to the structure prediction (e.g., Dynalign [25] and Carnac [26]); or (iii) the lowest free energy structure can be predicted individually for any sequence, and these ones are then aligned in order to find the structure shared by all of them (e.g., MARNA [27]). Comparative analysis, however, requires multi-alignment of available
homologous sequences, which presently make it not eligible approach for long and not conserved sequences of RNAs, instead of a single-sequence prediction analysis that is less demanding in this respect, but pays in term of a lower accuracy.
The choice between the different available tools has to be made according to the specific project aims. For example, for what concerns the lncRNAs [6,28,29], most the available tools are not immediately suitable to deal with them, due to the long sequence and the lack of multiple alignments of these RNAs.
We tackled this issue in our previous works [30,31], where we presented a novel pipeline called MONSTER (Method Of Non-branching Structures Extraction and search) that enables to detect structural motifs shared between two RNAs. MONSTER characterizes the RNA secondary structure through a descriptor-based method where the entire structure is made up of an array of more simple sub-structures (Figure 1). In particular, a predicted RNA secondary structure (Figure 1a) can be broken down into separated Non Branching Structures (NBSs, Figure 1b) that are conveniently represented by a dot-bracket notation (Figure 1c) [32]. Each NBS is described by an RNA Sequence-Structure Pattern (RSSP), i.e., a pair composed of a string of bases (the sub-sequence corresponding to the NBS) and a string that represents the secondary structure in the dot-bracket notation (the NBS). In addition, a list of parameters is associated to each RSSP and composes the header line. The set of RSSPs makes up the Secondary Structure Descriptor (SSD) of the RNA sequence (Figure 1d).
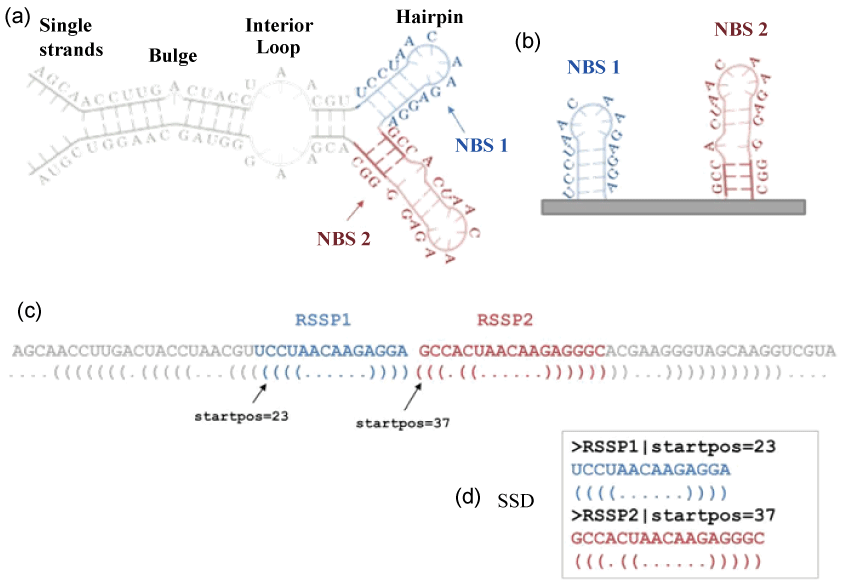
Figure 1: An example of the encoding of a predicted secondary structure into a Secondary Structure Descriptor (SSD). (a) RNA secondary structure representation with the two highlighted Non-Branching Structures (NBSs) (red one and blue one); (b) the extraction of the two NBSs; (c) mapping of the secondary structure in the dot-bracket notation (i.e., a 3-letter alphabet where dots represent unpaired bases, open-closed brackets "()" represent the paired bases) and the visualization of the two RSSPs that are a pair of the sub-sequence and the corresponding NBS; (d) the SSD composed of the two RSSPs.
View Figure 1
The underlying idea of MONSTER was to functionally characterize RNAs with unknown functions (target RNAs) by searching for similar structural motifs in RNA whose function is known (reference RNA). The prediction module of MONSTER makes use of RNALfold and thus comes under the methods that rely on single-sequence approach.
Here, we report a comprehensive comparison of two abovementioned approaches (i.e., single-sequence and comparative) with respect to the RNA structure predictions in terms of absolute and relative sensitivity of all the analyzed tools. Thus, we benchmark the prediction methods on a collection of RNA families with well-experimentally-known structures (e.g., making use of the freely-available database RNA strand v2.0) by comparing the predictions with respect to the experimentally-known structures.
Pursuing the idea of the RNA structure predictions comparison from several different tools, we developed two ad-hoc dynamic programming algorithms (SSD-opt and SSD-liberal), presented in the following, which are able to assess the accuracy of the most popular thermodynamic tool RNALfold from Vienna RNA package (Figure 2).
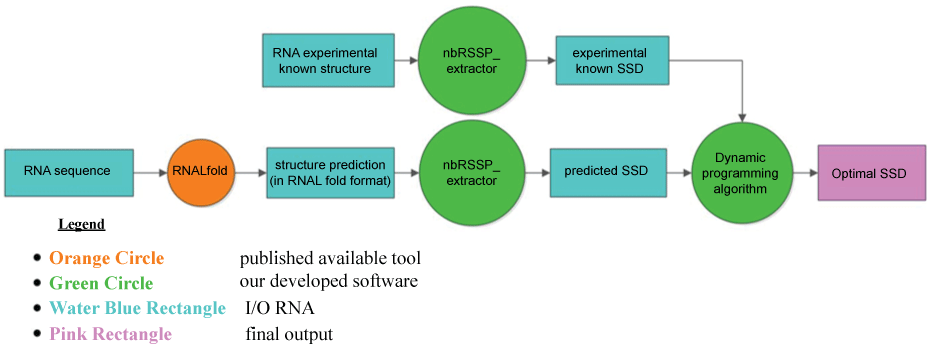
Figure 2: Flowcharts of the procedure built up to run SSD-opt or SSD-liberal. For each RNA input sequence, its local secondary structures are predicted by RNALfold; the corresponding overlapped NBSs are then extracted by the module nbRSSP-extractor of MONSTER [30,31] that returns the set of all RSSPs. Simultaneously, the known RNA structures are split in the corresponding true RSSPs by the module nbRSSP-extractor of MONSTER and the SSD of the known structures is obtained. Finally, pair wise comparisons are performed by SSD-opt or SSD-liberal between the predicted set of NBSs and the known structures. The output represents the optimal SSD. Legend: rectangles represent the Input/output blocks; the small circle represents the available published tool; the big circles represent our developed algorithms; the last rectangle on the right side represents the final output returned.
View Figure 2
RNALfold is a MFE-based predictor that returns the locally stable secondary structures of an RNA sequence according to a given parameter L that represents the maximum allowed distance between base-pairs. Additionally, it computes for each local structure its free energy, as well as the starting position in the sequence [21]. The output list is composed of all the possible local structures, which are predicted and may overlap (i.e., more predictions correspond to an identical piece of sequence).
Dynamic programming algorithms to evaluate accuracy of RNA structure predictions: SSD_opt and SSD_liberal
SSD_opt takes as input a set of predicted sub-structures (NBSs) and the related set of the experimentally-known ones (i.e., “Ground Truth”) and returns as output the array of non-overlapped predicted RSSPs that have the highest number of base-pairs matching with the experimentally-known ones. SSD_opt is based on dynamic programming, whose objective function is the maximization of the number of base-pairs according to what previously explain: it computes for each RSSP all the possible groups of RSSPs that have compatible starting positions and that begin with the same analyzed NBS. Finally, it returns the group of RSSPs whose computed score according the objective function is the optimal one. It is worth noting that in the case of two RSSPs having the same score, SSD_opt selects the one whose False Positive (FP) value (i.e., the number of predicted base-pairs that are not in the known RSSP) is lower.
Formally, we define: (i) R as the RNA sequence; (ii) S as the list of NBSs extracted from the predicted structure of R and sorted according to increasing sequence positions; (iii) T as the list of NBSs extracted from the experimentally-known structure of R; (iv) si the i-th NBS in S (i = 1,..,n with n = |S|) with pos(si) its position in R and length (si) its length; (v) ind(∙) as a function that can be applied to NBSs in S and returns the index of the argument in the list (starting from 1). Then, we consider as a chain of NBS in S such that satisfies the following conditions:
Specifically, condition (i) implies that C is sorted according to increasing positions in T, hence that To simplify notation, hereafter we will denote the matches in a chain as .
Based on these definitions, we define a function score to evaluate C as follows:
,
where P(si) is the number of base-pairs (i.e., pairs of brackets in the dot-bracket notation) of si that are also in T.
SSD-opt computes the score for all the chains of NBSs in S satisfying conditions (i) and (ii), and then selects the chain with the highest score. However, this is unfeasible for long sequences, since its complexity grows exponentially with the number of NBSs. To reduce the complexity, we consider for all s ∈ S only the chain ending with s that has the highest score. This can be done with dynamic programming using the recursion:
where
OPT(si) gives for any si ∈ S the highest score of chain ending with si and the corresponding optimal chain can be easily determined by backtracking. To conclude, SSD-opt taken as input S and T returns one optimal chain of NBSs in S.
SSD-liberal selects for each true NBS the predicted ones that have the highest number of base-pairs matching with the experimentally-known structures, regardless of any overlapping position. The algorithm takes as input a set of predicted NBSs (S) and the related set of the true ones (T). Thus, it returns as output the optimal chain of NBSs (even overlapped) based on the pair wise comparison of the predicted structure with the experimentally-known one.
Likewise SSD-opt, SSD-liberal is based on dynamic programming (see the previous subsection) and computes all the groups of RSSPs that begin with the same analyzed NBS and that reach the best score. However, instead of SSD-opt, the scores are assigned only by taking into account for the presence of each si ∈ S among the T list, without accounting for their overlapping positions. Therefore, the condition (ii) of SSD-opt has not to be satisfied. Indeed, in this case the recursive function of the dynamic programming algorithm is the following:
where
Methods for the RNA Structure Predictions
In this section, we list and describe the main algorithms able to predict and extract the secondary structure of both protein coding and non-coding RNAs.
Single-sequence methods
These methods predict the RNA secondary structure starting from the single sequence [30,33].
nbRSSP-extractor: nbRSSP-extractor [30] provides by default a unique prediction composed by non-overlapping RSSPs. Briefly, starting from a list of all possible (overlapped) local structures predicted by RNALfold (window size L = 150), nbRSSP_extractor extracts a set of NBSs that do not overlap, according to a specific selection criteria based on the means free energy per nucleotide. However, nbRSSP_extractor with a specific option can also return all the NBSs (even overlapped) that are extracted from RNALfold without any selection criteria (see RNALfold-lnrz method), as well as the NBSs contained in one unique global structure in the dot-bracket format (such as those NBSs extracted from the experimentally-known structure and that constitutes the list T).
RNALfold-lnrz: RNALfold-lnrz [30] analysis consists of applying the nbRSSP-extractor to select the non-overlapping predictions of RNALfold in an alternative way, i.e., the predictions of RNALfold are selected based on their decreasing free energies, and then the non-overlapping ones are chosen.
MFE-based: Based on the Free Energy Minimization (MFE), these methods start from the only single RNA sequence and determine the prediction of a secondary structure from thermodynamics. The aim is to find the base-pairing that provides the lowest free energy when a RNA molecule moves from the unfolded to the folded status. Mfold [11] and RNAfold [12] are based on the implementation of the Zuker-Stiegler algorithm to search for the lowest free energy structure by means of empirical estimations of the thermodynamics parameters. Finally, Fold algorithm (from the RNA structure package [17,18]) folds the RNA sequence into its lowest free energy conformation allowing the application of several constraints (e.g., modifications, required energy intervals, restrictions about the base-paring rules), as well as giving as output not only the lowest free energy structure, but all the possible ones.
ML-based: The software package Context Fold [14] relies on Machine-Learning (ML) techniques. It contains algorithms that provide a RNA structure prediction thanks to several scoring models that are trained on large training sets composed of RNA sequences with known structures.
MEA-based: Several methods are based on probabilistic approaches and look for the Maximum Expected Accuracy (MEA) structure in order to enlarge the information and effectiveness of their structure prediction. Among them, Sfold (sfold.wadsworth.org) performs a stochastic sampling of the structures given by the Boltzmann structures ensemble according to their occurring probability; then, it performs a clustering of the sampled structures. Centroid Fold predicts the RNA secondary structure improving their accuracy by means of generalized centroid estimators. Finally, iPknot (rtips.dna.bio.keio.ac.jp/ipknot/) predicts the MEA structure by using integer programming and accounting for the pseudoknots.
Comparative approaches
These methods predict the RNA secondary structure starting from multiple sequences in order to find the more conservative one (consensus structure) common to all (or almost all) the sequences [30,33].
Fold then align: This approach consists in predicting an array of structures having the lowest free energy for all the multiple sequences given as input. Then, it searches for the structure with lowest free energy shared among all the sequences.
An example of tools based on such an approach are MXScarna [34] (Multiplex Stem Candidate Aligner for RNAs) and MARNA [27]. MXScarna is a multiple alignment tool for RNA sequences that uses progressive alignment based on the pair wise structural alignment algorithm of SCARNA. MARNA is based on pair wise comparisons and it exploits the costs of the edit operations to compute the consensus structure of the input multiple alignments. To date, the most advanced LocARNA (bioinf.uni-freiburg.de/Software/LocARNA/) that performs a simultaneous alignment and folding replaced it.
Align then fold: Such an approach determines the multiple sequences alignment according to the RNA sequences information and then predicts the lowest free energy structure shared by the highest number of them. Centroid Alifold is based on the generalized centroid estimators to find the common lowest free energy structure. RNAalifold [12] implements an extension of the Zuker-Stiegler algorithm for computing consensus structures from RNA alignments. Finally, Pfold (daimi.au.dk/~compbio/pfold/) predicts the folding of an RNA alignment input by implementing a Stochastic Context Free Grammar, which is trained on a dataset of reference alignments.
Fold and align simultaneously: This approach makes use of Sankoff dynamic programming algorithm to simultaneously align and fold a set of RNA sequences [8,35]. Dyalingn implements a pairwise version of such an algorithm to identify a common lowest free energy structure and aligns two RNA sequences. Foldalign implements a local or global simultaneous folding and aligns two or more RNA sequences. Finally, Carnac implements an improved version of the Sankoff algorithm by adding several filters through which the set of sequences has to be processed. It calculates the base pairing probability matrices and aligns the sequences based on their full ensembles of structures.
Base pairing probability: The base pairing probability is defined as the probabilities of composing a base-pair in the ensemble of RNA secondary structures thanks to which the information about the single RNA structure can be enriched [23,36]. Among those tools that account for the base-pairing probabilities, Turbo fold of the RNA structure package [17] takes as input a set of homologous RNA sequences and folds them to identify the common structure with the lowest energy configuration. Specifically, it estimates the base pairing probabilities by intrinsic and extrinsic information to improve the accuracy of its RNA structure predictions. Furthermore, RNA sampler (stormo.wustl.edu/RNA Sampler) is a sampling-based program that includes structural pair wise information and base pairing probabilities estimation to predict common RNA secondary structure among multiple sequences. It is also able to deal with pseudoknots.
Evaluating the Performances of the RNA Prediction Tools
Here, we present the performances of some RNA folding algorithms on reliable and available data-sets of functional RNAs with experimentally-known secondary structures (e.g., rRNA 5S, 16S and 23S from RNAstrandv2.0 database, rnasoft.ca/sstrand). Thus, we compare the prediction results of the RNA folding algorithms with respect both to SSD-opt and SSD-liberal algorithms and to nbRSSP-extractor and RNALfold-lnrz performances, according to the metrics listed in table 1. The comparative analysis of the state-of-the-art tools have been rearranged from results reported in [10] and [33]. In particular, first we evaluate the performances of SSD-opt and SSD-liberal algorithms (Figure 3) with respect to the experimentally-known structures of the rRNAs families extracted from RNAstrandv2.0 database, and then we compare them with respect to the RNA secondary structure predictions of the other RNA folding algorithms.
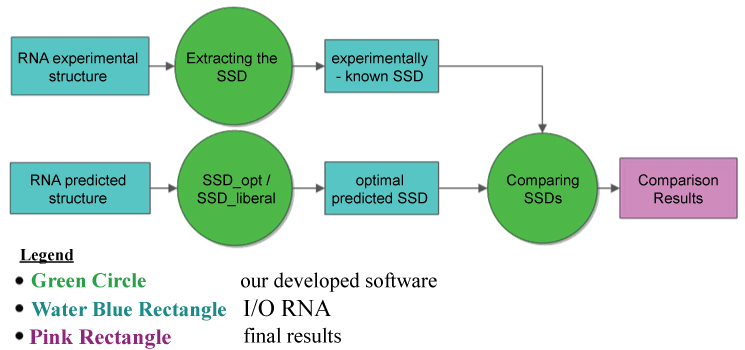
Figure 3: Flowcharts of the procedure to evaluate the SSD_opt (or SSD-liberal) performances. From each experimentally known RNA structure of 5s, 16s, 23s rRNA and tRNA families, the corresponding known Sequence Structure Descriptor is extracted (experimentally-known SSD). From each RNA families sequence the SSD-opt (or SSD-liberal) dynamics programming algorithm is applied in order to extract the corresponding optimal predicted SSD, by taking into account both the predicted structure as well as the known-experimental one (see Figure 2). Finally, pair wise comparisons are performed between the optimal predicted SSD and the experimentally-known ones. The procedure provides as output the comparison results with their computed statistics. Legend: rectangles represent the Input/output blocks; the big circles represent our developed algorithms; the last rectangle on the right side represents the final output returned.
View Figure 3
![]()
Table 1: Metrics.
View Table 1
We use the following metrics (Table 1) to measure the performances of all analyzed RNA structure prediction tools [37]:
1. TPR (True Positive Rate or Sensitivity): fraction of correctly predicted pairs of bases;
2. PPV (Positive Predictive Value): fraction of predicted base-pairs in the known structure;
3. F-measure: it is interpreted as a weighted harmonic mean of the sensitivity and PPV;
4. MCC (Matthew’s Correlation Coefficient): it can be approximated to the geometric mean between PPV and Sensitivity to evaluate the independence of prediction results between two algorithms.
TP (True Positive) values correspond to the correctly predicted base-pairs; TN (True Negative) values correspond to correctly unpaired predicted bases; FN (False Negative) values represents base-pairs that are in the reference true secondary structure but not in the predicted one; FP (False Positive) values correspond to base-pairs that are in the predicted structure but not in the reference one.
The performances of both SSD-opt and SSD-liberal algorithms are reported in details in table 2. In addition, we assess the comparison results of our novel implemented algorithms (SSD-opt and SSD-liberal) with respect to our previously developed ones (nbRSSP-extractor and RNALfold-lnrz), as well as with respect to the other state-of-the-art tools. These performance comparisons are reported in (Table 3).
![]()
Table 2: Results of SSD-liberal and SSD-opt algorithms on rRNA classes and tRNAs from the RNAstrand v2.0 database.
View Table 2
![]()
Table 3: Tool performance comparisons.
View Table 3
SSD-opt and SSD-liberal appear to reduce drastically the number of FP values (Table 2) and increase the TP ones with respect to the nbRSSP-extractor and RNALfold-lnrz analysis. In table 3, we can indeed observe as the TPR increases from the 0.56 value of nbRSSP-extractor up to the 0.66 value for SSD-opt and to 0.75 value for SSD-liberal.
Specifically, SSD-opt (Table 3) results at a comparable level in terms of TPR and PPV with respect to the other tools, while it shows higher performances in term of F-measure and MCC with respect to the single-sequence prediction tools (e.g., MFE-based, MEA-based [8], or ML-based [13]). For what concerns the comparison with respect to the comparative approaches, SSD-opt shows comparable results or lower ones in terms of PPV, although we have to underline that comparative methods often require sets of homologous sequences to perform the folding that are in some cases not available (e.g., lncRNAs). To conclude, the results of SSD-opt prove that RNALfold potentially enables to reach accurate predictions with lower computational costs with respect to other tools.
Furthermore, the results of SSD-liberal (Table 3) show as taking into account all the alternative predictions of RNALfold, we can reach a greater coverage of the possible matches between the predicted and experimentally-known structures. This is due to the following reasons: (i) on one hand, since SSD-liberal does not bind the search for the optimal SSD at the non-overlapped NBSs, it can perform it with a higher sensitivity; (ii) on the other hand, by using single-prediction tools, we compare a unique structure that does not means the better one. To this end, methods that account for alternative predictions could be represent a valid approach to enlarge the predictions sensitivity.
Conclusions
Here, we presented a comprehensive review of several approaches to the RNA structures prediction together with a detailed discussion of two novel algorithms (SSD-opt and SSD-liberal) that, starting from the local prediction of RNALfold, enable to efficiently find the optimal SSD of an RNA secondary structure based on the comparison with the well-characterized one. To test SSD-opt and SSD-liberal, we compare their performances with respect to these prediction tools on a collection of RNA families with well-experimentally-known structures. On one hand, the results obtained by SSD-opt show that RNALfold is potentially able to provide effective and accurate predictions. On the other hand, the performances of SSD-liberal reflect how methods that make use of alternative predictions enable to potentially enlarge the coverage of all the possible matches with the true structures. Therefore, a method that accounts for alternative predictions could be useful to address the RNA secondary prediction providing an increasing sensitivity, despite of a decreasing specificity.
Conflict of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Acknowledgments
G.F. acknowledges financial support from The Epigenomics Flagship Project EPIGEN funded by Italian Ministry of Education, University and Research (MIUR) and the National Research Council of Italy (CNR).
References
-
Mattick JS (2011) The central role of RNA in human development and cognition. Febs Letters 11: 1600-1616.
-
Eytan Zlotorynski (2015) Non-coding RNA: Circular RNAs promote transcription. Nature Reviews Molecular Cell Biology 16: 206.
-
Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, et al. (2010) Long Noncoding RNA as Modular Scaffold of Histone Modification Complexes. Science 329: 689-693.
-
Alessandro Fatica, Irene Bozzoni (2014) Long non-coding RNAs: new players in cell differentiation and development. Nature Reviews Genetics 15: 7-21.
-
Zhao Y, Qinhao Guo, Jiejing Chen, Jun Hu, Shuwei Wang, et al. (2014) Role of long non-coding RNA HULC in cell proliferation, apoptosis and tumor metastasis of gastric cancer: a clinical and in vitro investigation. Oncology reports 31: 358-364.
-
Victoria A Moran, Ranjan J Perera, Ahmad M Khalil (2012) Emerging functional and mechanistic paradigms of mammalian long non-coding RNAs. Nucleic Acids Res 40: 6391-6400.
-
Tim R Mercer, John S Mattick (2013) Structure and function of long noncoding RNAs in epigenetic regulation. Nature structural and molecular biology 20: 300-307.
-
Jan Gorodkin, Walter L. Ruzzo (2014) RNA Sequence, Structure, and Function: Computational and Bioinformatic Methods.: Humana Press, vol. 1097.
-
David H. Mathews, Douglas H. Turner (2006) Prediction of RNA secondary structure by free energy minimization. Current Opin in Struct Biol 16: 270-278.
-
Paul P Gardner, Robert Giegerich (2004) A comprehensive comparison of comparative RNA structure prediction approaches. BMC bioinformatics 5: 140.
-
Zucker M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406-3415.
-
Ronny Lorenz , Bernhart SH, Höner Zu Siederdissen C, Tafer H, Flamm C, et al. (2011) ViennaRNA Package 2.0. Algorithms for Molecular Biology 6: 26.
-
Haruka Yonemoto, Kiyoshi Asai, Michiaki Hamada (2015) A semi-supervised learning approach for RNA secondary structure prediction. Computational biology and chemistry 57: 72-79.
-
Zakov S, Goldberg Y, Elhadad M, Ziv-Ukelson M (2011) Rich parameterization improves RNA structure prediction. J Comput Biol 18: 1525-1542.
-
Ding Y, Lawrence CE (2003) A statistical sampling algorithm for RNA secondary structure prediction. Nucleic Acids Res 31: 7280-7301.
-
Zhi John Lu, Jason Gloor W, David H Mathews (2009) Improved RNA secondary structure prediction by maximizing expected pair accuracy. RNA 15: 1-9.
-
David H Mathews (2014) Using the RNAstructure Software Package to Predict Conserved RNA Structures. Current Protocols in Bioinformatics 46: 12-14.
-
Stanislav Bellaousov, Jessica S Reuter, Matthew G Seetin, David H Mathews (2013) RNAstructure: web servers for RNA secondary structure prediction and analysis. Nucleic Acids Res 41: 471-474.
-
Eugenio Mattei, Marco Pietrosanto, Fabrizio Ferrè, Manuela Helmer-Citterich (2015) Web-Beagle: a web server for the alignment of RNA secondary structures. Nucleic Acids Res 43: 493-497.
-
Eugenio Mattei, Gabriele Ausiello, Fabrizio Ferrè, Manuela Helmer-Citterich, (2014) A novel approach to represent and compare RNA secondary structures. Nucleic Acids Research 42: 6146-6157.
-
Hofacker I L, Priwitzer B, Stadler P F (2004) Prediction of Locally Stable RNA Secondary Structures for Genome-Wide Surveys. Bioinformatics 20: 186-190.
-
Sita J Lange, Maticzka D, Möhl M, Gagnon J N, Brown CM, et al. (2012) Global or local? Predicting secondary structure and accessibility in mRNAs. Nucleic Acids Res 40: 5215-5226.
-
Iwakiri J, Kameda T, Asai K, Hamada M (2013) Analysis of base-pairing probabilities of RNA molecules involved in protein-RNA interactions. Bioinformatics 29: 2524-2528.
-
Sean R Eddy (2014) Computational analysis of conserved RNA secondary structure in transcriptomes and genomes. Annual review of biophysics 43: 433-456.
-
Yinghan Fu, Gaurav Sharma, David H. Mathews (2014) Dynalign II: common secondary structure prediction for RNA homologs with domain insertions. Nucleic Acids Res 42: 13939-13948.
-
Touzet H, Olivie Perriquet (2004) CARNAC: folding families of related RNAs. Nucleic Acids Res 32: 142-145.
-
Sven Siebert, Rolf Backofen (2003) MARNA: A Server for Multiple Alignment of RNAs. GCB 135-140.
-
Sofia Quinodoz, Mitchell Guttman (2014) Long noncoding RNAs: an emerging link between gene regulation and nuclear organization. Trends in cell biology 24: 651-663.
-
M Morlando, M Ballarino, A Fatica, I Bozzoni (2014) The Role of Long Noncoding RNAs in the Epigenetic Control of Gene Expression. ChemMedChem 9: 505-510.
-
Giulia Fiscon, Paola Paci, Teresa Colombo, Giulio Iannello (2015) A new procedure to analyze RNA Non-branching Structures. BSP Current Bioinformatics 10: 242-258.
-
Giulia Fiscon, Paola Paci, Giulio Iannello (2015) MONSTER v1.1:a tool to extract and search for RNA non-branching structures. BMC Genomics 16: S1.
-
Meyer F, Kurtz S, Backofen R, Will S, Beckstette M (2011) Structator: fast index-based search for RNA sequence-structure patterns. BMC Bioinformatics 12: 214.
-
Puton T, Kozlowski L P, Rother K M, Bujnicki J M (2013) CompaRNA: a server for continuous benchmarking of automated methods for RNA secondary structure prediction. Nucleic Acids Res 41: 4307-4323.
-
Tabei, Yasuo, Kiyoshi Asai (2009) A local multiple alignment method for detection of non-coding RNA sequences. Bioinformatics 25:1498-1505.
-
Havgaard, Jakob Hull, Jan Gorodkin (2014) RNA structural alignments, part I: Sankoff-based approaches for structural alignments. Methods Mol Biol 1097: 275-290.
-
Kiyoshi Asai, Michiaki Hamada (2014) RNA structural alignments, part II: non-Sankoff approaches for structural alignments. Methods Mol Biol 1097: 291-301.
-
Xu Zhenjiang, Anthony Almudevar, David H Mathews (2012) Statistical evaluation of improvement in RNA secondary structure prediction. Nucleic Acids Res 40: 26.
