

Journal of Genetics and Genome Research
Sedaghatian-Type Spondylometaphyseal Dysplasia: A Case of Rapid Demise with Evidence of Myocardial Injury
Mehmet Sah İpek1* and Alper Akin2
1Departmentof Neonatology, Memorial Dicle Hospital, Diyarbakir, Turkey
2Department of Pediatric Cardiology, Dicle University Faculty of Medicine, Diyarbakir, Turkey
*Corresponding author:
Mehmet Sah İpek, Memorial Dicle Hospital, Urfa Road 3rd km, Kayapinar, Diyarbakir, Turkey, Tel: +904123156666, Fax: +904123156611, E-mail: md.msipek@yahoo.com
J Genet Genome Res, JGGR-3-025, (Volume 3, Issue 1), Case Report; ISSN: 2378-3648
Received: March 03, 2016 | Accepted: May 30, 2016 | Published: June 02, 2016
Citation: İpek MS, Akin A (2016) Sedaghatian-Type Spondylometaphyseal Dysplasia: A Case of Rapid Demise with Evidence of Myocardial Injury. J Genet Genome Res 3:025. 10.23937/2378-3648/1410025
Copyright: © 2016 İpek MS, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Sedaghatian-type spondylometaphyseal dysplasia (SSMD) is a lethal neonatal form of spondylometaphyseal dysplasias that is reported rarely. The case of a female infant demonstrating a clinical phenotype consistent with a diagnosis of SSMD was presented. She died with acute evidence of myocardial injury.
Conclusion: In SSMD, certain organ systems including the central nervous, cardiac, and skeletal systems, are clearly involved. Although the exact cause of the uniform fatality remains unclear, cardiac involvement seems to play a role in the rapid and unexpected death.
Keywords
Sedaghatian-type spondylometaphyseal dysplasia, Myocardial injury, Lethal, Neonate
Introduction
Sedaghatian-type spondylometaphyseal dysplasia [SSMD; OMIM 250220] is a lethal neonatal form of spondylometaphyseal dysplasia characterized by severe metaphyseal chondrodysplasia with mild limb shortening, platyspondyly, cardiac conduction defects, and central nervous system (CNS) abnormalities [1-6]. Sedaghatian was first described in 1980 in three siblings of non consanguineous Iranian parents. A total of 19 infants from 13 families have been reported to have SSMD [2]. Many of the infants with SSMD have been reported to have a short life span, dying in the first few days of life due to respiratory failure [1], with the longest surviving infant living to 161 days [4]. Cardiac abnormalities [1-7] and CNS malformations [1,3,4,7] have been reported frequently in SSMD. Herein, we present a new case of a female infant demonstrating a clinical phenotype consistent with a diagnosis of SSMD and resulting in rapid demise with evidence of myocardial injury.
Case Report
A full-term newborn female infant was referred to a neonatal intensive care unit with respiratory distress that developed soon after birth as well as a congenital anomaly. She was a product of cesarean section delivery to a gravida 12, para 6, abortus 6, live birth 5 mother, and she had an Apgar score of 8 and 9 at 1 and 5 minutes, respectively. Her mother was 31-years-old and pregnant from a consanguineous marriage. Although there were no regular antenatal check-ups, antenatal ventriculomegaly was detected at 32 weeks of gestation. The family history indicated that a sibling had previously died within the first week of life.
On admission, the infant was hypoactive and hypotonic, with a blood pressure of 64/40 mmHg, pulse rate of 154 beats/min, respiration rate of 72 breaths/min, body temperature of 36.8°C, and oxygen saturation of 90% on room air. On physical examination, her length was 46 cm (25th-50th centile), and her head circumference was 36 cm (50th-75th centile). She exhibited rhizomelic shortening of all four limbs, and redundant skin folds were noted in her limbs (Figure 1a). Her chest and hands appeared normal. The laboratory findings including initial arterial blood gases as well as biochemical and hematological investigations were unremarkable. Skeletal survey revealed increased intervertebral disc spaces, platyspondyly, pronounced cupping of the medial and lateral ends of the ribs, short long bones, irregularity, widening, and cupping of the metaphyses of long tubular bones in the upper and lower extremities, small hypoplastic iliac bones, horizontal acetabular roofs with pronounced medial spurs and small sacrosciatic notches, disproportionately long fibulae, and shortening and cupping of short tubular bones (Figure 1b). Electrocardiography (ECG), echocardiography, and abdominal ultrasound were normal on the first day of admission. Computerized tomographic images of the cranium showed colpocephaly, enlarged ventricles, a simplified gyral pattern, and agenesis of the corpus callosum. Cranial ultrasound scan also confirmed an absent corpus callosum (Figure 2). Unfortunately, brain magnetic resonance imaging could not be performed.
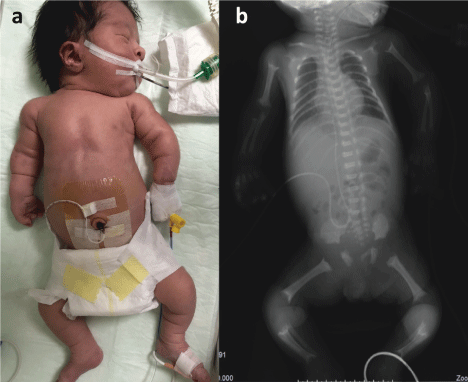
Figure 1: General appearance (a) and radiograph (b) of case. Note that rhizomelic shortening of all four limbs, and redundant skin folds in the limbs (a). Short long bones, widening and cupping of metaphyses of all bones in the extremities, platyspondyly, ribs cupped anteriorly and posteriorly, flat acetabular roof, decreased sacrosciatic notch, small hypoplastic iliac bones, and disproportionately long fibulae are seen on radiograph (b).
View Figure 1
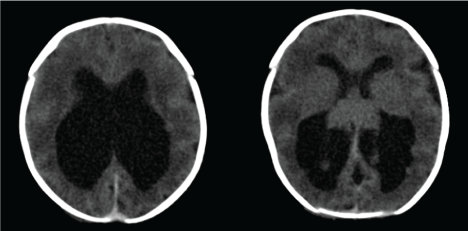
Figure 2: Computerized tomography of cranium showing colpocephaly, enlarged ventricles, and pachygyria.
View Figure 2
The infant was monitored, provided oxygen supplement, and commenced on nasogastric feeding. On the second day, she suddenly deteriorated and required intubation, volume resuscitation, and inotropic support. Arterial blood gas analysis was consistent with metabolic acidosis. During clinical deterioration there was a moderately depressed left ventricule ejection fraction (35%) and mild repeated mitral regurgitation on echocardiography. Unfortunately, ECG was not available during clinical deterioration. However, no cardiac arrhythmia was detected by continuous monitoring.
Despite all interventions, the infant died at the third hour of deterioration. The retrospective analyses of the sera, which were taken for biochemical analysis before deterioration, revealed that troponin-I and creatine kinase myocardial band (CKMB) were 25 ng/ml (Roche Diagnostics, RotkreuzGmbH, Switzerland, cobas e 601; normal value < 0.5 ng/ml) and 521 ng/ml (Roche Diagnostics, Rotkreuz GmbH, Switzerland, cobas c501; normal value < 25 ng/ml), respectively. The parents refused an autopsy.
Discussion
SSMD is a rare, lethal disorder characterized by normal intrauterine growth, neonatal respiratory distress/insufficiency, rhizomelic shortening of the long bones, brachydactyly, redundant skin folds, somewhat narrow but otherwise normal rib cage, cardiac conduction defects, metaphyseal cupping and irregularity, platyspondyly, delayed epiphyseal ossification, and irregular iliac crests [1-3,8]. The radiographic findings are characteristic, including metaphy seal cupping, flaring, and irregularity, cupped ribs, platyspondyly, irregular ossifications in iliac wings and calcaneus, short metacarpals and phalanges, and disproportionately long fibulae [1,8]. The radiologic and clinical features in the present case are consistent with a diagnosis of SSMD.
CNS malformations are consistent with common abnormal neuronal migration, including severe cerebellar hypoplasia, partial lissencephaly, frontotemporal pachygyria, porencephaly, agenesis of the corpus callosum, and simplified gyral pattern, all of which have been reported in cases of SSMD [1,3,4]. These findings may be responsible for the hypotonicity, convulsions, and respiratory failure observed in patients with SSMD [1].
Cardiac conduction disturbances including complete heart block have been frequently reported in SSMD [1,5,6]. Some nonlethal structural cardiac anomalies have also been reported [1-4]. It has been speculated that some affected babies with complete heart block may have remained undiagnosed due to very early neonatal death [1]. In the case presented here, the ECG taken on the first day was normal; unfortunately, it was not available during clinical deterioration. Although some previously reported neonatal deaths were due to cardiac arrhythmia [5,6], such a relationship could not be documented in our case. However, the findings of echocardiography and elevated levels of myocardial markers indicated myocardial injury, which may have resulted from myocarditis or myocardial ischemia. Myocardial markers were not evaluated in previously reported cases, but subacute myocarditis was described among the autopsy findings in one case [9]. There are various causes of troponin level elevation [10], but evidence for those conditions was not detected in this case. Because the sera for myocardial markers were taken before clinical deterioration, we suggest that myocardial injury was not secondary to metabolic acidosis, which developed later. It is also noted that serum troponin-I concentration was much higher in our case compared with previously reported fatal asphyxiated neonates (21 ng/ml vs. 12.8 ng/ml) [11].
The parental relationship in our case further supports autosomal recessive inheritance, as previously considered [1,5,6,9]. However, due to insufficient information, it is not clear whether the sibling of the deceased was similarly affected. It has recently been shown that recessive truncating mutations in the glutathione peroxidase 4 gene in two families affected with SSMD could be a cause of this lethal form of spondylometaphyseal dysplasia [3]. Moreover, congenital malformations in the CNS, cardiac, and skeletal systems suggest the possibility of lipid peroxidation inhibition [3].
In conclusion, skeletal dysplasia is characteristic of SSMD, but it does not appear to be severe enough to predict a rapidly fatal outcome, and many other organ systems are clearly involved. In the case presented here, the cause of rapid and unexpected clinical deterioration may be myocardial injury. However, the mechanisms of myocardial injury and the exact cause of the uniform fatality remain unexplained. Future studies should seek to clarify the importance of lipid peroxidation in SSMD pathogenesis.
Contributors
MSİ: management of case; MSİ, AA: wrote the manuscript; AA: supervision and final approval of manuscript.
Informed Consent
Parent consent was obtained for publishing the baby's photo.
Funding
The authors have no support or funding to report.
Conflict of Interest
The authors declare that they have no conflict of interest.
References
-
Aygun C, Celik FC, Nural MS, Azak E, Kucukoduk S, et al. (2012) Simplified gyral pattern with cerebellar hypoplasia in Sedaghatian type spondylometaphyseal dysplasia: a clinical report and review of the literature. Am J Med Genet A 158A: 1400-1405.
-
Sedaghatian MR (1980) Congenital lethal metaphyseal chondrodysplasia: a newly recognized complex autosomal recessive disorder. Am J Med Genet 6: 269-674.
-
Smith AC, Mears AJ, Bunker R, Ahmed A, MacKenzie M, et al. (2014) Mutations in the enzyme glutathione peroxidase 4 cause Sedaghatian-type spondylometaphyseal dysplasia. J Med Genet 51: 470-474.
-
Koutouby A, Habibullah J, Moinuddin FA (2000) Spondylometaphyseal dysplasia: Sedaghatian type. Am J Med Genet 90: 199-202.
-
Kerr B, Smith V, Patel R, Ladusans E, Sillence DO (2000) Spondylometaphyseal dysplasia Sedaghatian type associated with lethal arrhythmia and normal intrauterine growth in three siblings. Clin Dysmorphol 9: 167-172.
-
Mahendran SM, Wilcox FL, Chirumamila L (2007) Spondylometaphyseal dysplasia-Sedaghatian type associated with intra-partum cardiac arrhythmia and neonatal death. J Obstet Gynaecol 27:851-853.
-
Campbell RS, Ireland M, Bloxham CA, Chippindale AJ (1992) Platyspondylic lethal osteochondrodysplasia: Shiraz type with radiological-pathological correlation. Pediatr Radiol 22: 90-92.
-
Elcioglu N, Hall CM (1998) Spondylometaphyseal dysplasia-Sedaghatian type. Am J Med Genet 76: 410-414.
-
Opitz JM, Spranger JW, Stöss HR, Pesch HJ, Azadeh B (1987) Sedaghatian congenital lethal metaphyseal chondrodysplasia-Observations in a second Iranian family and histopathological studies. Am J Med Genet 26: 583-590.
-
Correale M, Nunno L, Ieva R, Rinaldi M, Maffei G, et al. (2009) Troponin in newborns and pediatric patients. Cardiovasc Hematol Agents Med Chem 7: 270-278.
-
Türker G, Babaoglu K, Gökalp AS, Sarper N, Zengin E, et al. (2004) Cord blood cardiac troponin I as an early predictor of short-term outcome in perinatal hypoxia. Biol Neonate 86: 131-137.
