

Journal of Hypertension and Management
Treatment of Idiopathic Pulmonary Arterial Hypertension: Current and Clinical Trial Modalities
Raheel Khan, Taimur Abbasi, Roberto F. Machado and Jiwang Chen*
Section of Pulmonary, Critical Care, Sleep and Allergy, Department of Medicine, University of Illinois at Chicago, USA
*Corresponding author:
Jiwang Chen, PhD, Section of Pulmonary, Critical Care, Sleep and Allergy, Department of Medicine, University of Illinois, 909 South Wolcott Avenue, M/C 719, Chicago, IL 60612, USA, Tel: 312-355-1470, Fax: 312-996-7193, E-mail: chenjw@uic.edu
J Hypertens Manag, JHM-2-005, (Volume 2, Issue 1), Review Article; ISSN: 2474-3690
Received: October 10, 2015 | Accepted: February 29, 2016 | Published: March 02, 2016
Citation: Khan R, Abbasi T, Machado RF, Chen J (2016) Treatment of Idiopathic Pulmonary Arterial Hypertension: Current and Clinical Trial Modalities. J Hypertens Manag 2:005. 10.23937/2474-3690/1510005
Copyright: © 2016 Khan R, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Pulmonary hypertension (PH) is a deadly cardiovascular disease that follows a progressive and symptomatic course. This disease presents in approximately 15 cases per million each year and is categorized into five different groups based on established or presumed etiology. The poorly understood role of BMPR II mutation, early onset and severity of symptoms and increased fatality rate associated with IPAH make treatment a challenge for physicians. Current treatment options focus on the three main mechanisms of endothelial dysfunction which is the hallmark of PAH, namely Prostacyclin, Endothelin-1, and Nitric oxide pathways. This review summarizes different mechanisms of action for drugs that are currently available, recently approved or still under investigation. Despite all the progress made in the past decade, a number of obstacles still exist in the way of optimal treatments and outcomes. Currently available drugs (Amlodipine, Ambrisentan, and Sildenafil) may no longer be as effective as the newer generation of drugs, due to better outcomes and favorable modification of disease profile. Some clinical trial drugs have shown significant health improvements in IPAH patients by prolonging vasodilation and inhibiting vasoconstriction. Understanding the mechanism of action for IPAH still remains the key for researchers and physicians to develop novel drugs to treat PAH patients thereby increasing survival rates.
Keywords
Pulmonary arterial hypertension, Treatment, Clinical trials, Drug
Introduction
Pulmonary hypertension (PH) is a debilitating and progressive disease which results in an increase in the pulmonary vascular resistance (PVR), right heart failure, and death if left untreated [1-3]. The continual increase in blood pressure negatively affects pulmonary arteries, leading to pulmonary arterial hypertension (PAH) [4,5]. The mean pulmonary artery pressure (mPAP) in normal adults is between 10-18 mmHg, but in patients with PAH the mPAP values are ≥ 25 mmHg at rest and ≥ 30 mmHg during exercise; the wedge pressure for PAH < 15 mmHg. The continual increase of mPAP ≥ 45 mmHg at rest is called severe PAH [6]. The survival rates for patients with mPAP ≥ 42.5 mmHg are significantly lower than similar groups of patients with a mPAP < 42.5 mmHg; the one and five year survival rates are 82% and 57%, respectively [7,8]. The drastic reduction in survival rates is due to right ventricular heart failure resulting from PAH.
While PH encompasses five different categories, group one PAH is of utmost concern to clinical researchers and physicians because of the increased fatality rates and underlying ambiguity [2,8-11]. According to the U.S. registry of patients, among people with PH nearly 6% had a first degree relative that suffered from the same disease. Recognition for familial transmission of the disease was evident in the early 2000’s when allelic variants of the bone morphogenetic protein receptor (BMPR2) gene were found to be associated with hereditary PH. The BMPR2 gene is located on chromosome 2 and this variant triggers changes in the amino acid makeup of BMPR2 protein resulting in impaired signal transmission during the mechanics of pulmonary arterial smooth muscle cell (PASMC) apoptosis, leading to unbridled cellular proliferation [12].
It has been shown that nearly 65% of families with hereditary PAH have various allelic variants in both the intron and exon regions of the BMPR2 gene. These allelic variants are usually transmitted in an autosomal dominant fashion, whereas a considerable number of spontaneous incidents of IPAH involve novel exon allelic variants. The penetrance of the allelic variants is low, having a mean of nearly 20% and is variable among families, essentially a person with BMPR2 allelic variant has only a 1 in 5 chance of developing PAH, and the offspring of people carrying the variant gene has a 1 in 10 of developing PAH. This lethal mutation increases the mPAP, consequently leading to right heart failure. Because group one PAH encompasses scleroderma and idiopathic/familial causes, identifying the origin of the disease of IPAH has gained significant clinical attention. The golden standard for identifying PAH is through right heart catherization. Because physicians are not certain of the underlying cause of group one PAH, they resort to treating symptoms rather than treating the disease - such as using beta blockers for high blood pressure or vasodilators for narrowed arteries [13].
Further mutations which facilitate the proliferation of PH include mutations in the activin-like kinase type 1 TGF-beta receptor which is located on chromosome 12 at the ACVRL1 gene. This receptor is located on endothelial cells. Variants in the allele for ACVRL1 have been implicated in patients having both PAH and hereditary hemorrhagic telangiectasia (HHT). Allelic variants in the endoglin (ENG) located on chromosome 9 have also been implicated in patients having both these pathologies, HTT and PAH.
Proliferation of PASMC is controlled by a number of various elements, one of them being 5-Hydroxytryptamine transporter (5-HTT). An allelic variant in a gene promoter associated with increased expression of 5-HTT has also been implicated in IPAH. This understanding of allelic variants and polymorphisms promises to be the vanguard of future treatments and a source of improvement in currently available regiments.
Regardless of causes and associations, certain interrelated mechanisms play a role in the biopathologic evolution and mechanism of IPAH. These mechanisms are chaperoned by a number of molecular and cellular processes, which includes paucity of prostacyclin caused by reduced synthase activity, elevated levels of endothelin from increased production and lowered clearance, decreased expression of nitric oxide synthase, low levels of platelet 5-HTT and increased plasma levels of 5-HTT, down regulation of various voltage-gated potassium channel activity of pro-inflammatory cytokines, unbridled activity of various autoantibodies pro-thrombotic states from various pathological dysfunctions involving platelets, and decoagulation cascades. All these conditions lead to a predisposition of the pulmonary vasculature to steer towards vasoconstriction rather than vasodilation partly due to the fact that cellular proliferation supersedes cellular apoptosis leading to a state of thrombotic diathesis.
As the vascular pathology progresses inducing adventitial fibrosis, medial hypertrophy, and/or intimal thickening, pulmonary vascular resistance (PVR) increases and mPAP rises in order to maintain a constant level of cardiac output (CO) (Figure 1). As the right ventricle (RV) compensates for the resistance, the mPAP will increase as long as the PVR increases. The increased RV pressure will eventually lead to right heart failure and the mPAP will decline as the patient decompensates. Failure to maintain constant CO levels eventually leads to death (Figure 1).
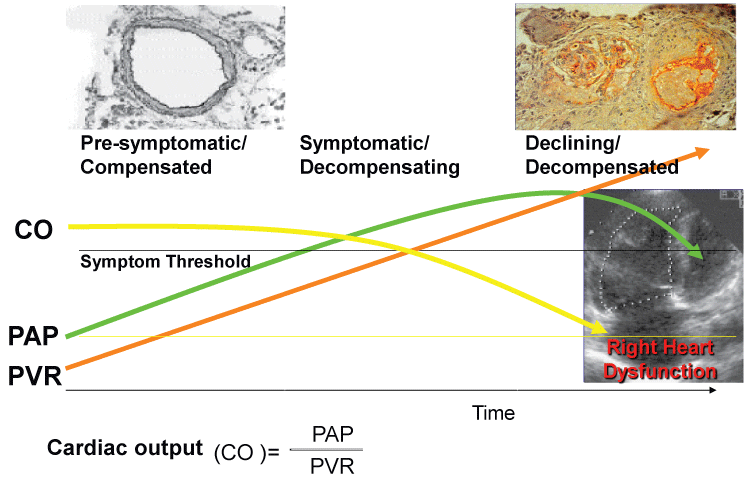
.
Figure 1: Schematic progression of pulmonary arterial hypertension (PAH).
PAP: Pulmonary artery pressure; PVR: Pulmonary vascular resistance.
View Figure 1
In general, it is proposed that because of certain susceptibilities such as genetic, anatomic, or drug related various individuals are more prone to developing pulmonary arterial vasoconstriction, which is an early harbinger of PAH. For the past 2 decades the focus of novel therapy development has been focused on the three main pathways of endothelial dysfunction [14]. This review will focus on the current and clinical trial treatment options - highlighting the mechanism of action for both the currently available and clinical trial drugs for IPAH therapy. We mainly focused on pre-2010 drug options because the vast majority of the common PAH modalities used in treatments were approved before 2010; otherwise this demarcation was arbitrary and merely simplified our focus of review. We also reviewed the recently approved drugs including Macitenan, Riociguat and Orentram, which were developed in 2013. By highlighting the mechanism of action for currently available drugs such as Ambrisentan, Amlodipine, and Epoprostenol, the way in which these drugs promote vasodilation or inhibit vasoconstriction can be identified. In addition, by understanding the currently available drugs and comparing these drugs to potential routes of administration, we were able to review Riociguat, Selexipag and Ralinepag in an effort to understand how these drugs work similarly or differently to the currently available drugs and the overall effect that these drugs had on PAH patients.
Current Medication Targeted Pathways
Most of the common current treatment drugs/inhibitors are presented in Table 1 and Table 2. Each drug category is specific in the way it interacts with smooth muscle cells of the arteries and/or the cardiac muscle cells.
![]()
Table 1: Drugs currently available (post-2010) or under clinical trials for PAH.
View Table 1
![]()
Table 2: Drug inhibitors for the 5 different mechanisms of action.
View Table 2
Calcium channel blockers, CCB, are a group of drugs in which Ca2+ entry into excitable cells is inhibited. By preventing Ca2+ influx into the coronary and peripheral arterial smooth muscles and the heart, Ca2+ will not serve as an intracellular messenger [15]. Consequently, CCBs serve as a smooth-muscle dilator in addition to having a negative inotropic, negative chronotropic, and negative dromotropic effect on the cardiac cells of the atria and ventricles as well as an effect on the smooth-muscle layers of the pulmonary arteries and veins (Table 1). By dissolving into the phospholipid bilayer, CCBs will interact with the binding site in smooth muscle cells thereby promoting vasodilation. Amlodipine, a dihydropyridine calcium antagonist, inhibits calcium from crossing into the phospholipid bilayer thus preventing Ca2+ from entering into the vascular smooth muscle and cardiac muscle cells. Previous studies have been shown that Amlodipine binds to the dihydropyridine at the non-dihydropyridine binding sites. When Amlodipine binds to the binding sites, it relaxes the arterial muscles and dilates the pulmonary and coronary arteries/veins. When the blood vessels dilate, this allows the heart to have a greater CO; thereby reducing the blood pressure and increasing oxygen and nutrient distribution throughout the body [16]. CCBs are limited for PAH patients who show a response to acute vasoreactivity tests, the intended use cannot be used generally for all PH related cases, thus requiring further advancements in clinical research for PAH-related drugs.
Prostanoids are a class of drugs that are synthetically derived from the prostate glands. Prostanoids are important in maintaining vascular homeostasis because these drugs inhibit platelet aggregation, vasoconstriction, and smooth muscle proliferation. One common prostanoid is Epoprostenol [17]. When Epoprostenol binds at the prostacyclin receptor, this lowers mPAP, PVR, and increases CO. When Epoprostenol is administered to PAH patients > 40 ng/kg/min, the mPAP value decreases by 8 mmHg and the survival rate with prolonged Epoprostenol use is 1-3 years. Chronic infusion of Epoprostenol is delivered via a small portable pump through a central venous catheter. With chronic use, the 6 minute walking distance, 6MWD, increases by 60 meters within 12 weeks. The 6MWD is a test in which the patient walks at a comfortable pace for the longest distance he/she can, the walking distance is calculated and measured based on the age, sex, and type of cardiovascular disease; the average healthy adult has a 6MWD between 400-700 meters. While Epoprostenol has shown marked improvements in the 6MWD, it constituted a ceiling effect, which an individual can only show a limited improvement. For example, a patient can show an increase of 50 meters in the 6MWD in the first 3 weeks, but as the patient is continuously administered Epoprostenol, there will no longer be an increase in the 6MWD. This plateau is because the body can only tolerate a specific amount of the given drug and will compensate, the binding sites and receptors will become continually exposed to the drug [17].
One of the most common prostanoids for IPAH is Iloprost, a second generation structural analog of prostacyclin (PGI) with a roughly ten-fold greater potency than the first generation stable analogs, such as carbaprostacyclin. This drug was approved by FDA in 2004, and has been on the market since then. Iloprost binds with equal affinity to human prostacyclin (Prostanoid IP) and prostaglandin EP1 receptors. Iloprost constricts the ilium and fundus circular smooth muscle as strongly as prostaglandin E2 (PGE2) itself and inhibits the ADP, thrombin, and collagen-induced aggregation of human platelets. Iloprost acts as a vasodilator and this helps to antagonize the pathological changes that take place in the small pulmonary arteries of patients with pulmonary hypertension [18].
Endothelian receptor antagonists (ERA) are a class of drugs that inhibit the binding of an Endothelian vaso-constrictive peptide to its receptors on smooth muscle cells, resulting in vasodilation. Studies have shown that PAH patients have shown improvements in exercise and a slower disease progression when given an ERA. Endothelian-1 is a 21 amino acid oligopeptide which acts in a paracrine and autocrine fashion as a vasoconstrictor [19]. This oligopeptide stimulates the neighboring cell which leads to blood vessel constriction. There are two different ERA receptors, ERA-A and ERB. ERA-A is a receptor found on smooth muscle cells, and ERB is a receptor found on vascular endothelial cells. When an Endothelian receptor antagonist blocks the Endothelian receptors, this limits harmful excess Endothelian in the blood vessels. When the ERA receptor is blocked, this prevents vasoconstriction of blood vessels. However, when the ERB receptor is blocked, this inhibits vasodilation. Consequently, the ERB receptor is believed to be a counter-regulatory mechanism: it protects against vasoconstriction, and allows greater cardiac output. There are two types of inhibitors, the first blocks both ERA and ERB and the second inhibitor only blocks the ERA receptor.
Bosentan, a FDA approved drugs in 2001, is a dual endothelin receptor antagonist important in the treatment of PAH. Bosentan blocks the endothelin receptors and substrates that would otherwise promote narrowing of the blood vessels leading to high blood pressure. Patients with PAH have elevated levels of endothelin, a potent blood vessel constrictor, in their plasma and lung tissue. Endothelin-1 (ET-1) is a neurohormone, the effects of which are mediated by binding to ETA and ETB receptors in the endothelium and vascular smooth muscle. ET-1 concentrations are elevated in plasma and lung tissue of patients with pulmonary arterial hypertension, suggesting a pathogenic role for ET-1 in this disease. Bosentan has a slightly higher affinity for ETA receptors than for ETB receptors, thus promoting vasodilation [20]. This vasoconstriction inhibition and vasodilation promotion alleviates PAH symptoms, i.e. high blood pressure, smaller cardiac output, and angina.
Phosphodiesterase-5 (PDE5) inhibitors promote vasodilation in blood vessels in the lungs; this is done by preventing the breakdown of cyclic guanosine monophosphate (cGMP) [21]. Mediators such as nitric oxide (NO) prolong vasodilation. Vasodilation in pulmonary arteries/veins allows for more blood to become oxygenated. A PDE5 inhibitor prevents the hydrolysis of cGMP vasoconstrictor, thereby promoting continuous vasodilation. When there is an abundance of cGMP in the cells, this mediates smooth muscle relaxation (Table 1). When cGMP levels become low, the nitric oxide activates guanylyl cyclase, thereby dephosphorylating GTP to cGMP. When cGMP is absent, smooth muscles constrict. By having an abundance of cGMP, due to GTP phosphorylation, this prevents vasoconstriction. A PDE5 inhibitor, Sildenafil, prevents the hydrolysis and degradation of cGMP and allows continuous smooth muscle relaxation, thereby promoting vasodilation. With continuous smooth muscle relaxation, PAH patients experience a gradual decrease in blood pressure, angina absence, and a greater cardiac output. All these results lead to an increase in oxygen and nutrient distribution.
Soluble guanylyl cyclase (sGC) stimulants are drugs which have similar actions to PDE5 inhibitors [22]. The sGC is an enzyme which interacts in the nitric oxide cycle; this enzyme promotes the binding of NO to a heme group on the sGC. When this occurs, the downstream mechanism allows for the catalysis and synthesis of cGMP. When cGMP is produced, this allows for smooth muscle vasodilation- until cGMP is hydrolyzed (Table 1). By increasing the affinity and the number of sGC enzymes, this allows for greater cGMP production and prolonged vasodilation, in accordance with the PDE5 inhibitor. sGC can be either a stimulator or an activator, stimulators inform sGC to low levels of NO for the NO cycle, and then they stabilize a nitrosyl-heme complex and maintain the enzyme in the active configuration (Table 1). The new sGC stimulants work by acting as a receptor for the nitric oxide. Once the nitric oxide binds to the sGC, the combined enzyme catalyzes the synthesis of cGMP. Once cGMP is produced, it promotes vasodilation in the pulmonary blood vessels. Vasodilation will allow more blood flow and nutrient flow to the rest of the body; this will reduce the blood pressure and help oxygenate the body’s tissues.
Recently Approved Drugs
Macitentan, a relatively new ERA; was FDA approved in 2013 after completing the phase III SERAPHIN study. Macitentan works in a similar way as most ERA’s, by binding to one or both of the Endothelian receptors [23]. By only binding to receptor A, this promotes vasodilation. Conversely if only receptor B is bound, this promotes vasoconstriction. The SERAPHIN study was a multicenter, double-blind, placebo-controlled, parallel-group, event driven, Phase III outcome study in 742 patients with symptomatic PAH who were randomized to three treatment groups (placebo [N = 250], 3 mg [N = 250] or 10 mg [N = 242] of Macitentan once daily), to assess the long-term effect on morbidity or mortality. Treatment with Macitentan 10 mg resulted in a 45% risk reduction of the composite morbidity-mortality endpoint up to EOT when compared to placebo [24].
The treatment effect was established early and was sustained. In patients already on background therapy for PAH treatment with Macitentan 10 mg resulted in 38% risk reduction of the composite morbidity-mortality endpoint compared to placebo. The risk of PAH related death or hospitalization for PAH up to EOT was reduced by 50% in patients receiving Macitentan 10 mg (50 events) compared to placebo [25]. Macitentan works by preventing the binding of ET-1 to ERA-A and ERB. This is because Macitentan displays a high affinity and occupancy of the ET receptor in human pulmonary arterial smooth endothelial muscle cells. In addition to this high affinity, Macitentan also has a higher affinity to bind to ERA-A than to ERB and because there are more ERA-A receptors than ERB receptors, this promotes vasodilation and consequently inhibits vasoconstriction [25-27].
Riociguat was FDA approved on October 8, 2013. This relatively new drug is a PDE5 inhibitor; this drug prevents the hydrolysis of cGMP. cGMP presence promotes smooth muscle relaxation which will eventually promote vasodilation. This drug underwent a PATENT-1 phase III clinical trial in 2013. PATENT-1 is a double-blind, randomized, placebo-controlled trial of 443 patients with PAH at 124 centers in 30 countries. Patients were randomly assigned in 2:4:1 ratio to; placebo, Riociguat in individually adjusted doses up to 2.5 mg three times daily (2.5 mg maximum group), or Riociguat in individually adjusted doses that were capped at 1.5 mg three times daily (1.5 mg maximum group). The primary endpoint was a change from baseline to the end of week 12 in the 6-minute walk distance (6MWD). At week 12, 6MWD had increased from baseline by a mean of 30 m in the 2.5 mg-maximum group and had decreased by a mean of 6 m in the placebo group (least-squares mean difference, 36 m; 95% confidence interval: 20 to 52; P < 0.001). Significant benefits were seen in the 2.5 mg-maximum group, as compared with the placebo group, with respect to a range of secondary end points including pulmonary vascular resistance (P < 0.001) [28-31].
Notably, patients who were receiving endothelin-receptor antagonists or non-intravenous prostanoids were permitted into the study and accordingly half of patients were on background therapy for PAH: 44% with endothelin-receptor antagonists and 6% with non-intravenous prostanoids. Pre-specified subgroup analysis showed that Riociguat improved the 6MWD in patients who had not received other PAH-targeted therapies and also in those who had been on endothelin-receptor antagonists or prostanoids [28]. This phase III clinical trial data illustrates the importance of the overall outcome of Riociguat - the marked decrease in PAH symptoms and the marked increase in the 6MWD.
Orentram was FDA approved in 2013 for PAH patients. This drug, a treprostinil, acts as a selective IP-prostacyclin receptor agonist. This drug will promote vasodilation and inhibit vascular smooth muscle cells proliferation. This drug is currently available for oral administration in 0.125 mg, 0.25 mg, 1 mg and 2.5 mg dosage tablets. FDA approval of Orenitram was based on the results obtained from a phase III clinical trial, known as FREEDOM-M study. It was a randomized, double blind, and parallel assignment, which enrolled 349 PAH patients. The study was conducted between October 2006 and April 2011. The primary outcome measure of the study was the six-minute walk distance (6MWD) from baseline to week 12. The secondary outcome measures included the 6MWD from base line to weeks eleven, eight and six, and clinical worsening assessment. The study results demonstrated that the patients treated with Orenitram in a twice-daily dose showed improvement in the median six-minute walk distance (6MWD) by +23m when compared with patients receiving only the placebo [32-34].
Another phase III clinical trial, known as FREEDOM-C, was also conducted on Orenitram, across 72 centers worldwide, between October 2006 and December 2010. The randomized, multi-center, double blind and parallel assignment enrolled 354 PAH patients. The primary outcome measure of the study was the 6MWD from baseline to week 16 [32].
Clinical Trial Drugs
Clinical trial drugs have shown marked improvements in phase I and phase II trials, these innovative drugs are also showing greater health improvements due to their new emergence and ability to bind to ERA receptors or due to inhibition of cGMP hydrolysis. While CCBs, prostanoids, ERAs, PDE5 inhibitors, and sGC stimulators are all effective drugs on the market now; due to their prolonged use and because of the constant change in diseases- these drugs will not be as effective as they once were [12]. For example, drugs such as Amlodipine and Epoprostenol were approved by the FDA in 1999 and 1995, respectively; these drugs have been on the market for 20 years and prolonged exposure led to a ceiling effect. Clinical trial drugs are potentially more effective on combating the early onset symptoms of PAH and the deleterious effects of the BMPR II mutation because of the new ways in which these drugs dilate blood vessels or affect the IP receptors/ERA binding sites. Current drugs combated the illness in ways that were not previously attempted for PAH patients, such as preventing Ca2+ influx or prolonged vasodilation due to cGMP accumulation, after 20 years of use, these drugs are not as effective as they once were.
Selexipag is a drug which specifically targets the prostacyclin pathway, Prostacyclin receptor [35]. Prostanoids are compounds which primarily act on signaling substances and directly communicate with the central nervous system and the control mechanisms for immune and inflammatory responses. Prostacyclin promotes vasodilation, acts as an anti-inflammatory, and inhibits platelet accumulation. If the prostacyclin cycle is impaired, it can promote vasoconstriction and inflammation. These symptoms are commonly seen in PAH patients, possibly due to the excess endothelial cells. Selexipag works by acting as a selective IP-prostacyclin receptor agonist. Because prostacyclin activates the IP-receptor; this allows for vasodilation and inhibition of proliferation of vascular smooth muscle cells. Selexiag underwent a phase III GRIPHON study, was used by 1,156 patients with PAH; reported on June 2014 by Actelion Pharmaceuticals, Ltd. As the Selexipag was titrated up, there were signs of decreased risks of morbidity/mortality as compared to the placebo. This result was a double-blind experiment where the dose of Selexipag was uptitrated from 200 µg twice a day to 1600 µg twice a day, for 4.2 years. These results proved that Selexipag is a significant IP-receptor and PAH treatment option, based on the 39% (p < 0.0001) significance factor. However, in greater than 5% of Selexipag clinical trial participants - these participants did experience adverse side effects: headache, diarrhea, jaw pain, nausea, myalgia, vomiting, pain in extremity, and flushing [35].
Ralinepag is another prostacyclin IP-receptor, similar to Selexipag [36]. This new drug is still undergoing phase II clinical trials at the University of San Diego Medical School. This drug, like most Prostacyclin receptors, slows the progression of PAH as well as improves the tolerance to exercise; i.e. the 6MWD. In phase 1 of the clinical trial, Ralinepag underwent a randomized, double-blind, placebo-controlled study to evaluate its safety, tolerability, and pharmacokinetics. This drug was administered as 0.03 mg, 0.05 mg, 0.1 mg, and 0.2 mg single doses. This up-titration allowed researchers to find the tolerance level for each of the thirty-two research participants. The participants were randomly assigned to one of the eight groups, each group had four participants. Among the eight groups, six of the groups were given Ralinepag while the other two were given a placebo. After dose administration and data analysis, Ralinepag moved to phase 2, including sixty patients with PAH. Currently these patients are being titrated to their appropriate tolerance level, over the course of nine weeks. For the remaining thirteen weeks, the patients will be given the same tolerance dosage of the drug [36].
Of the newer novel agents, only Selexipag targets the disease via the conventional pathway of prostacyclin receptors; other novel agents under study take aim at the disease through other not yet completely understood mechanisms [37,38].
The role of growth factors such as platelet derived growth factor (PDGF), Fibroblast growth factor (FGF), Epidermal growth factor (EGF) and their interaction with Tyrosine Kinase receptors (RTK) in the pathogenesis of PH via the proliferation and migration of endothelial cells and vascular smooth muscle cells has been described. Thus the inhibition of these RTK receptors has been the focus of various therapies. Of these Tyrosine kinase receptor inhibitors (TKI) is Imatinib, it inhibits the BCR-ABL receptor, that in various animal studies had shown to modify pulmonary vascular smooth muscle remodeling and improve survival. However clinical trials in humans have been beset with various hurdles and beg further investigation into the efficacy of the drug [39].
The vasoconstriction, remodeling and proliferation of the pulmonary vascular smooth muscles is also mediated via the Rho kinase GTPases (RhoA & ROCKS), thus presenting another potential avenue for novel therapy development. Work on inhibitory pathways in animal models has been promising. Experimental therapies in this class include Fasudil, and Aziandole-1 both of which are ROCK inhibitors and have shown to reduce PVR and MPAP in various limited studies with human patients [40-44].
Serotonin (5-HT) is an important biological vasoconstrictor, the over expression of the serotonin receptor has been studied in PAH. Various studies have shown that, while not as a monotherapy but when used in concert with ERAs, the serotonin receptor antagonists showed a favorable effect when measured as a decrease in PVR or improvements in 6MWD test. The antagonist used was Terguride [45,46]. However much more studies need to be conducted before this can be claimed conclusively as a potential treatment augment, especially to use synergistically with other modalities that have received ceiling effect.
Prospect
In an effort to develop therapies for PAH, physicians need to further research and understand the underlying mechanism of PAH. By treating the mechanism of action, physicians will be more equipped in treating the underlying cause of the disease. There are constantly new clinical trial drugs emerging on the market such as; UT-15 and bordoxolone methyl. These drugs might result in PAH symptom reduction, but they will not completely eliminate PAH, unless they work on the molecular level and inhibit PAH progression. PAH primarily affects one of the three pathways: Prostacyclin pathway, Endothelian pathway, or the PDE-5 pathway. Drugs specific to these different pathways all provide a similar outcome for PAH patients, vasodilation. In combination with the emergence of these new classes of drugs and the previously successful drugs (Epoprostenol, Macitentan, Riociguat), PAH can be aggressively treated by targeting multiple effector pathways; this will provide symptomatic and increased survival benefits to the PAH patients. Now that physicians understand the BMPR II mutation, they are able to prescribe the appropriate dosage in an effort to compensate for the de novo exon mutation or heritable mutations. The differences in drug distribution and the way these drugs interact differently for each individual is a task the physician faces when dealing with PAH. This personalized medicine will be more effective than giving a standard treatment of care for each individual. By taking advantage of the preclinical data and understanding PAH, physicians will be more equipped in studying the underlying mechanism for IPAH and creating treatment options that will not only treat the symptoms but prevent the progression of the disease.
In addition to understanding the mechanism by which PAH is acquired and the way different drugs can combat PAH symptoms, it is equally important to maintain a healthy lifestyle. By maintaining a healthy eating habit and through daily exercise, this will significantly reduce early PAH onset symptoms. An increase in mPAP can result from many things, an increase PVR or even an increase in sodium consumption [47-50]. By monitoring the daily sodium consumption, not to exceed 2000 mg, the chance of increased mPAP is significantly reduced. Another way as to limit PAH progression is to reduce red meat intake. By decreasing red meat intake and limiting the amount of trimethylamine-N-oxide (TMAO) consumed, this will lower the significant role in the pathogenesis of hypertension. By reducing carnitine consumption, this will alleviate high blood pressure by lowering the levels of TMAO in hypertensive patients. While new drugs and dietary changes are being constantly updated, these changes are meaningless unless individuals educate themselves. By educating the public of the potential causes of PAH and preventing the occurrence and progression of PAH, one severely limits the chance of right heart failure and PAH. By providing a greater emphasis on dietary changes and increasing exercise, the prevalence of cardiovascular diseases such as PAH will greatly reduce.
It is imperative to understand the genetic background of PAH. By understanding the most lethal form of PAH, IPAH associated with the BMPR II mutation, he/she will be more proactive in looking for ways to reduce PAH. The connection between BMPR II mutations and early onset symptoms of PAH is important because of the increased fatality due to heritable or de novo mutations. While most PAH patients do not have a BMPR II mutation, these mutations are most common among PAH patients, approximately 70% of cases. Even though 70% of PAH patients have the BMPR II mutation, only 30-40% of those individuals will experience the adverse effects, the remaining patients will be carriers of this allelic mutation [25]. While the BMPR II mutation is most common in PAH patients, it is still prevalent in groups two through five.
Despite a few ceiling effects, clinical trial drugs, recently approved drugs, dietary changes, and understanding the genetics behind IPAH provided a notable increase in the 6MWD, lowering of arterial blood pressure, and a marked decrease in PAH symptoms. Scientists are still using different strategies in developing and testing these new drugs. These new emerging drugs are most commonly tested on pre-clinical animal models of pulmonary hypertension. The early stages of research into IPAH shows significant promise and will greatly help combat adverse health problems PAH patients’ experience.
Acknowledgement
This study was supported in part by grants from the Pulmonary Hypertension Association Proof-of-Concept Research Grant (to J.C.).
References
-
Miravitlles M, Price D, Rabe KF, Schmidt H, Metzdorf N, et al. (2015) Comorbidities of patients in tiotropium clinical trials: comparison with observational studies of patients with chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis 10: 549-564.
-
Gidwani S, Nair A (2014) The burden of pulmonary hypertension in resource-limited settings. Glob Heart 9: 297-310.
-
Lan B, Hayama E, Kawaguchi N, Furutani Y, Nakanishi T (2015) Therapeutic efficacy of valproic acid in a combined monocrotaline and chronic hypoxia rat model of severe pulmonary hypertension. PLoS One 10: e0117211.
-
Lohani O, Colvin KL, Yeager ME (2015) Biomarkers for pediatric pulmonary arterial hypertension: challenges and recommendations. Paediatr Respir Rev 16: 225-231.
-
Klabunde Richard E (2011) Cardiovascular Physiology Concepts. (2nd edn), Lippincott Williams & Wilkins, 6-9.
-
Raja SG, Raja SM (2011) Treating pulmonary arterial hypertension: current treatments and future prospects. Ther Adv Chronic Dis 2: 359-370.
-
Baker WL, Radojevic J, Gluck JA (2015) Systematic Review of Phosphodiesterase-5 Inhibitor Use in Right Ventricular Failure Following Left Ventricular Assist Device Implantation. Artif Organs 40: 123-128.
-
Vaidya B, Gupta V (2015) Novel therapeutic approaches for pulmonary arterial hypertension: Unique molecular targets to site-specific drug delivery. J Control Release 211: 118-33.
-
Liang OD, Mitsialis SA, Chang MS, Vergadi E, Lee C, et al. (2011) Mesenchymal Stromal Cells Expressing Heme Oxygenase-1 Reverse Pulmonary Hypertension. Stem Cells 29: 99-107.
-
Low AT, Medford AR, Millar AB, Tulloh RM (2015) Lung function in pulmonary hypertension. Respir Med 109: 1244-1249.
-
Ghofrani HA, Staehler G, Grünig E, Halank M, Mitrovic V, et al. (2015) Acute effects of riociguat in borderline or manifest pulmonary hypertension associated with chronic obstructive pulmonary disease. Pulm Circ 5: 296-304.
-
Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, et al. (2009) Updated clinical classification of pulmonary hypertension. Turk Kardiyol Dern Ars 42: 45-54.
-
Ryan JJ, Huston J, Kutty S, Hatton ND, Bowman L, et al. (2015) Right ventricular adaptation and failure in pulmonary arterial hypertension. Can J Cardiol 31: 391-406.
-
Pulido T, Zayas N, de Mendieta MA, Plascencia K, Escobar J (2016) Medical therapies for pulmonary arterial hypertension. Heart Fail Rev .
-
Rogers NM, Seeger F, Garcin ED, Roberts DD, Isenberg JS (2014) Regulation of soluble guanylate cyclase by matricellular thrombospondins: implications for blood flow. Front Physiol 5: 134.
-
Ying Z, Giachini FR, Tostes RC, Webb RC (2009) Salicylates dilate blood vessels through inhibiting PYK2-mediated RhoA/Rho-kinase activation. Cardiovasc Res 83: 155-162.
-
Saito Y, Nakamura K, Akagi S, Sarashina T, Ejiri K, et al. (2015) Epoprostenol sodium for treatment of pulmonary arterial hypertension. Vasc Health Risk Manag 11: 265-270.
-
http://www.drugbank.ca/drugs/DB01088
-
Kochegarov AA (2003) Pharmacological modulators of voltage-gated calcium channels and their therapeutical application. Cell Calcium 33: 145-162.
-
http://www.drugbank.ca/drugs/DB00559
-
Shanmugam E, Jena A, George M (2015) Riociguat: Something new in pulmonary hypertension therapeutics? J Pharmacol Pharmacother 6: 3-6.
-
Morrell NW (2006) Pulmonary hypertension due to BMPR2 mutation: a new paradigm for tissue remodeling? Proc Am Thorac Soc 3: 680-686.
-
Paul, Jean (2014) Selexipag. Acetlion, 1: 1-2.
-
Paul, Jean (2014) Macitentan. Acetlion 1: 1-3.
-
Sidharta NP, Treiber A, Dingemanse J (2015) Clinical Pharmacokinetics and Pharmacodynamics of the Endothelin Receptor Antagonist Macitentan. Clin Pharmacokinet 54: 457-461.
-
Khadka A, Sing Brashier DB, Tejus A, Sharma AK (2015) Macitentan: An important addition to the treatment of pulmonary arterial hypertension. J Pharmacol Pharmacother 6: 53-57.
-
Tonelli RA, Ahmed KM, Alkukhun L, Cikach F, Aulak K, et al. (2015) Treprostinil Iontophoresis in Idiopathic Pulmonary Arterial Hypertension. Am J Respir Crit Care Med 192: 1014-1016.
-
Said K (2014) Riociguat: PATENT-1 Study. Glob Cardiol Sci Pract 2014: 31-35.
-
Garnock-Jones KP (2014) Riociguat: A review of Its Use in Patients with Chronic Thromboembolic Pulmonary Hypertension or Pulmonary Arterial Hypertension. Drugs 74: 2065-2078.
-
Khaybullina D, Patel A, Zerilli T (2014) Riociguat (Adempas): a Novel Agent For the Treatment of Pulmonary Arterial Hypertension and Chronic Thromboembolic Pulmonary Hypertension. Pharmacy and Therapeutics P&T 39: 749-758.
-
Sulica R, Fenton R, Cefali F (2015) Early Observations on the Use of Riociguat in a Large, Metropolitan Pulmonary Arterial Hypertension/Chronic Thromboembolic Pulmonary Hypertension Treatment Center. Cardiol Ther 4: 209-218.
-
(2015) Orenitram (treprostinil) for Treatment of Pulmonary Arterial Hypertension (PAH). Drug Development Technology, Kable.
-
Gleason BJ, Dolan J, Piran P, Rahaghi FF (2015) The Rapid Initiation, Titration, and Transition from Intravenous to Oral Treprostinil in a Patient with Severe Pulmonary Arterial Hypertension. Case Rep Pulmonol: 498981.
-
Preston RI, Feldman J, White J, Franco V, Ishizawar D, et al. (2014) Safety and efficacy of transition from inhaled treprostinil to parenteral treprostinil in selected patients with pulmonary arterial hypertension. Pulm Circ 4: 456-459.
-
Audet, Craig M (2015) Arena Pharmaceuticals Initiates Phase 2 Clinical Trial Evaluating Ralinepage for Pulmonary Arterial Hypertension. Arena 1: 1-2.
-
Klabunde, Richard E (2011) Cardiovascular Physiology Concepts. (2nd edn), Lippincott Williams & Wilkins, 1-3.
-
Galie N, Humbert M (2015) ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. The joint task force for the diagnosis and treatment of pulmonary hypertension of the European society of cardiology (ESC) and the European respiratory society (ERS). Eur Respir J. 46: 903-975.
-
Simonneau G, Torbicki A, Hoeper MM, Delcroix M, Karlócai K, et al. (2012) Selexipag: an oral, selective prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. Eur Respir J 40: 874-880.
-
Montani D, Bergot E, Günther S, Savale L, Bergeron A, et al. (2012) Pulmonary arterial hypertension in patients treated by dasatinib. Circulation 125: 2128-2137.
-
Liao JK, Seto M, Noma K (2007) Rho kinase (ROCK) inhibitors. J Cardiovasc Pharmacol 50: 17-24.
-
Atallah E, Durand JB, Kantarjian H, Cortes J (2007) Congestive heart failure is a rare event in patients receiving imatinib therapy. Blood 110: 1233-1237.
-
Fukumoto Y, Matoba T, Ito A, Tanaka H, Kishi T, et al (2005) Acute vasodilator effects of a Rho-kinase inhibitor, fasudil, in patients with severe pulmonary hypertension. Heart 91: 391-392.
-
Fujita H, Fukumoto Y, Saji K, Sugimura K, Demachi J, et al. (2010) Acute vasodilator effects of inhaled fasudil, a specific Rho-kinase inhibitor, in patients with pulmonary arterial hypertension. Heart Vessels 25: 144-149.
-
Pankey EA, Byun RJ, Smith WB 2nd, Bhartiya M, Bueno FR, et al (2012) The Rho kinase inhibitor azaindole-1 has long-acting vasodilator activity in the pulmonary vascular bed of the intact chest rat. Can J Physiol Pharmacol 90: 825-835.
-
Guignabert C, Raffestin B, Benferhat R, Raoul W, Zadigue P, et al (2005) Serotonin transporter inhibition prevents and reverses monocrotaline-induced pulmonary hypertension in rats. Circulation 111: 2812-2819.
-
Dumitrascu R, Kulcke C, Königshoff M, Kouri F, Yang X, et al. (2011) Terguride ameliorates monocrotaline-induced pulmonary hypertension in rats. Eur Respir J 37: 1104-1118.
-
Ogawa A, Ejiri K, Matsubara H (2014) Long-term patient survival with idiopathic/heritable pulmonary arterial hypertension treated at a single center in Japan. Life Sci. 118: 414-419.
-
Gonzalez F, Liu B, Machado RF, Chen (2015) Does red meat induce hypertension? Austin Journal of Pulmonary and Respiratory Medicine 2: 1019.
-
Reddy V, Sridhar A, Machado RF, Chen J (2015) High sodium causes hypertension: evidence from clinical trials and animal experiments. J Integr Med 13: 1-8.
-
Ryu SH, Kim JW, Kim YS, Lee SH, Cho YB, et al. (2014) Negligible pharmacokinetic interaction of red ginseng and antihypertensive agent amlodipine in Sprague-Dawley rats. J Toxicol Environ Health A. 77: 1372-1383.
-
Lang GM, Paterno R, Faraci MF, Heistad DD (1997) Mechanisms of Adrenomedullin-Induced Dilation of Cerebral Arterioles. Stroke 28: 181-185.
-
Hinson PJ, Kapas S, Smith MD (2000) Adrenomedullin, a Multifunctional Regulatory Peptide. Endocr Rev 21: 138-167.
-
Hamid R, Newman JH (2009) Evidence for inflammatory signaling in idiopathic pulmonary artery hypertension: TRPC6 and nuclear factor-kappaB. Circulation 119: 2297-2298.
-
Maier T, Follmann M, Hessler G, Kleemann HW, Hachtel S, et al. (2015) Discovery and pharmacological characterization of a novel potent inhibitor of diacylglycol-sensitive TRPC cation channels. Br J Pharmacol 172: 3650-3655.
-
Archer SL, Weir EK, Wilkins MR (2010) Basic science of pulmonary arterial hypertension for clinicians: new concepts and experimental therapies. Circulation 121: 2045-2066.
-
Ramiro-Diaz JM, Nitta CH, Maston LD, Codianni S, Giermakowska W, et al. (2013) NFAT is required for spontaneous pulmonary hypertension in superoxide dismutase 1 knockout mice. Am J Physiol Lung Cell Mol Physiol 304: L613-625.
-
Minatuski S, Miura I, Yao A, Abe H, Muroaka H, et al. (2015) Pletelet-Derived Growth Factor Receptor-Tyrosine Kinase Inhibitor, Imatinib, is Effective for Treating Pulmonary Hypertension Induced by Pulmonary Tumor Thrombotic Microangiopathy. Int Heart J 56: 245-248.
