

Journal of Hypertension and Management
Cardiac Fibrosis in Hypertension
Tracey Keteepe-Arachi and Sanjay Sharma*
Department of Cardiovascular Sciences, St George's University of London, UK
*Corresponding author:
Sanjay Sharma, MD, Department of Cardiovascular Sciences, St George's University of London, Cranmer Terrace, London, UK, Tel: +442087255939, Fax: +442082979100, E-mail: sasharma@sgul.ac.uk
J Hypertens Manag, JHM-3-023, (Volume 3, Issue 1), Review Article; ISSN: 2474-3690
Received: October 22, 2016 | Accepted: February 11, 2017 | Published: February 14, 2017
Citation: Keteepe-Arachi T, Sharma S (2017) Cardiac Fibrosis in Hypertension. J Hypertens Manag 3:023. 10.23937/2474-3690/1510023
Copyright: © 2017 Keteepe-Arachi T, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Myocardial fibrosis is the hallmark of myocardial remodelling found in hypertensive individuals. This process adversely affects the outcomes of such patients and results in diastolic and systolic cardiac dysfunction, electrical dysrhythmia and potentially sudden death. Collagen metabolism has been highlighted as the primary mechanism by which fibrosis is regulated. However, there are many other facets to the pathophysiology of myocardial fibrosis including mechanical, cellular and hormonal influences, which may guide therapy and thereby determine prognosis.
Introduction
Hypertension remains one of the most significant modifiable causes of morbidity and mortality in the UK. The global prevalence of hypertension in the year 2000 was almost one billion and is set to increase by 29% to 1.56 billion by 2025, making the condition a major public health issue [1,2]. Hypertensive heart disease (HHD) represents a constellation of sequelae including left ventricular hypertrophy (LVH), diastolic dysfunction, heart failure and arrhythmia. Myocardial fibrosis, commonly associated with ischaemic scar formation, is an important phenomenon in the development of HHD.
Pathophysiology
Normal myocardium consists of cardiomyocytes and extracellular matrix which is composed of fibrillar collagen. Cardiac adaptation in response to hypertension is a complex and multifaceted process involving cardiomyocyte hypertrophy, fibroblast activation and extracellular matrix expansion. Mechanisms by which myocardial fibrosis occurs are being elucidated; both haemodynamic and neurohormonal, and various circulating biomarkers have been identified. In addition, collagen metabolism, which is fundamental to the development of myocardial fibrosis, can be evaluated by using markers of collagen turnover [3,4].
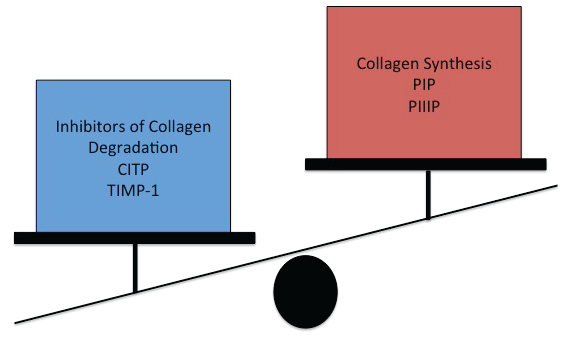
In hypertensive heart disease an excess of collagen is found in the extracellular matrix of the myocardium. This is thought to be due to an imbalance in collagen metabolism resulting from both increased synthesis as well as normal or reduced degradation [5,6]. Measurements of collagen turnover are divided into markers of collagen synthesis [carboxy-terminal propeptide of procollagen type I (PIP), carboxy-terminal propeptide of procollagen type III (PIIIP)], markers of collagen degradation [carboxy-terminal telopeptide of collagen type I (CITP)], markers of inhibitors of collagen degradation [tissue inhibitor of matrix metalloproteinases type I (TIMP-1)] and markers of fibroblast activity [transforming growth factor β1 (TGF β1)] [7] (Figure 1).
.
Figure 1: Markers of collagen metabolism.
PIP = carboxy-terminal propetide of procollagen type, PIIIP = carboxy-terminal propetide of procollagen type III, CITP = carboxy-terminal telopeptide of collagen type I, TIMP-1 = tissue inhibitor of matrix metalloproteinases type I.
View Figure 1
These markers are altered in hypertensive patients [8]. Many studies have demonstrated that in hypertensive individuals with LVH, serum PIP was found in greater quantities than in normotensive counterparts [5,9,10]. In addition these raised levels of PIP corresponded to increased collagen volume fraction (CVF) found on endomyocardial biopsy as well as a reduction in levels corresponding to regression secondary to antihypertensives [11]. In hypertensive individuals with echocardiographic signs of diastolic dysfunction a significant increase in TIMP I was demonstrated when compared to normal controls [12].
Upregulation of the procollagen type I gene and reduced collagenase activity have been demonstrated in experimental research and animal studies [13,14].
Mechanical factors such as pressure overload affecting the left ventricle, are implicated in triggering increased collagen synthesis and reduced collagenase activity [15]. Concomitant myocyte hypertrophy further alters myocardial architecture.
Morphological features secondary to hypertension and an increase in CVF include reactive interstitial fibrosis, perivascular fibrosis and reparative fibrosis. The thickening of pre-existing collagen fibres and the appearance of an increased number of collagen fibres is termed reactive fibrosis. Perivascular fibrosis indicates collagen accumulation within the adventitia of arterioles and arteries. Reparative fibrosis occurs because cardiac myocytes are not able to replicate or regenerate as they are terminally differentiated, and so fibrillar collagen replaces lost myocytes after necrosis, forming areas of scar [6]. Due to adaptation to myocyte necrosis and subsequent loss of parenchymal tissue, reparative fibrosis acts to maintain structural integrity of cardiac tissue.
Left Ventricular Hypertrophy
Hypertension is a pivotal factor in the development of left ventricular hypertrophy (LVH), which occurs as a compensatory mechanism to reduce wall stress. Increased after load causes cardiomyocyte hypertrophy, fibroblast stimulation and collagen synthesis [16].
Myocardial remodelling ensues with a resultant increase in fibrous tissue formation. Athletes also exhibit LVH yet do not suffer from cardiac dysfunction implying that the presence of LVH is not the primary determinant, but the degree of myocardial fibrosis. Physiological LVH represents the normal response to exercise and is a mechanism by which cardiac output matches demand which is not the case in pathological LVH [17].
The presence of LVH, detected by electrocardiogram or echocardiogram is a strong independent risk factor for cardiovascular morbidity and mortality. The repercussions of hypertension and LVH include development of atrial fibrillation and potentially fatal ventricular arrhythmia and chronic heart failure [18]. Concentric LVH is a milestone on the journey towards heart failure. Excluding age, LVH is the main predictor of adverse cardiovascular outcomes in hypertensive patients. It is an independent risk factor for heart failure, cerebrovascular disease, coronary heart disease and sudden death [2]. Importantly, intervention with antihypertensive treatment has been shown to reduce the risk of potential adverse cardiovascular events in these patients due to their beneficial effects on left ventricular mass [19,20].
Patterns of Left Ventricular Hypertrophy
The increased LV mass response to raised blood pressure is varied among individuals. For example, significant racial variability exists, with blacks exhibiting greater increases in LV mass and worse diastolic dysfunction when exposed to comparable levels of hypertension in contrast to white counterparts [21,22].
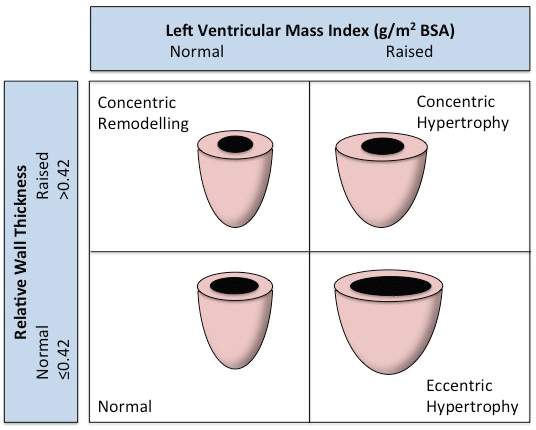
In hypertensive patients, LV mass is increased due to left ventricular wall thickening and/or chamber dilatation [23]. These two patterns of response have been classified according to echocardiographic criteria using relative wall thickness (RWT) as the variable parameter. RWT is calculated as 2x posterior wall thickness/LV diastolic diameter. When RWT is above 0.42, LVH is termed concentric. LVH is termed eccentric when RWT is not greater than this value. Concentric remodelling which is a third pattern of LVH is present when RWT is increased but LV mass is not. Patients with hypertension may have any of these patterns of LVH as demonstrated by multiple echocardiographic studies (Figure 2).
.
Figure 2: Diagrammatic representation of the classification of left ventricular hypertrophy.
View Figure 2
In a study of 3042 individuals with normal LV function echocardiography was used to evaluate systolic function and LVM at baseline and at 5-years. Interestingly, concentric LVH typically seen in hypertensive heart disease, was not shown to be a risk factor for LV impairment. In contrast eccentric LVH was found to be a predictor for development of reduced LVEF independent of coronary artery disease and MI [24]. It remains to be seen as to whether eccentric LVH may be a prognostic marker for those hypertensive patients at risk of developing a more deleterious LVH phenotype which results in systolic LV dysfunction. This suggests a relationship between geometric patterns of remodelling and cardiovascular outcomes and mortality.
Haemodynamic Precipitants of Left Ventricular Hypertrophy
In hypertensive individuals systolic blood pressure is the main determinant of LVH, which occurs as a direct result of increased wall stress due to increased cardiac workload. Left ventricular hypertrophy serves to relieve wall tension and thus preserve systolic ventricular function. Stimulation of procollagen gene expression, collagen protein synthesis, collagen deposition and ultimately fibrosis are the consequences of chronic pressure overload [3,25]. When exposed to mechanical load, cardiac fibroblasts demonstrate an increase in collagen type I synthesis and a reduction in collagenase expression [15]. The importance of the pressure load mechanism is demonstrated by a study by Verdecchia, et al. where hypertensive patients were divided into two groups: usual control (SBP < 140 mmHg) versus tight control (SBP < 130 mmHg) and the rate of electrocardiographic LVH was assessed at 2 year follow up. The tight control group had a rate of 11.4% and the usual control group a rate of 17% (p = 0.013) suggesting that progression to LVH could be reduced with more rigorous control of systolic blood pressure [26].
Both in vitro and in vivo experiments have demonstrated that mechanical load influences collagen metabolism. The collagen volume fraction (CVF) has been shown to be significantly increased in biopsy samples and post mortem specimens of patients with hypertensive heart disease compared to samples from normotensive individuals [27].
The effects of transmural gradients of wall stress are highlighted by a study by Tanaka, et al. which indicated an increase in CVF from the outer to the inner third of the left ventricular wall [28]. Querejeta, et al. also observed that in patients with HHD, systolic blood pressure and pulse pressure were proportional to CVF [10].
Non-Haemodynamic Precipitants of Left Ventricular Hypertrophy
Evidence suggests that biological variables in addition to mechanical factors, contribute significantly to myocardial fibrosis. In animal models, angiotensin II has been identified as a potential mechanism, independent of blood pressure, in the development of cardiac fibrosis in hypertensive disease [29]. It causes fibroblast proliferation, changes in collagen turnover, mRNA expression and type I collagen synthesis is also increased with subsequent stimulation of aldosterone and deposition of collagen type I and III fibres and resultant fibrosis [30]. In addition, angiotensin II attenuates the proteolytic activity of collagenase causing progressive collagen accumulation leading to deformation of myocardial tissue and an increase in myocardial stiffness, diastolic dysfunction and eventually systolic dysfunction.
The role of the renin-angiotensin-aldosterone system (RAAS) in the development of cardiac fibrosis is highlighted by a number of experimental studies. In unilateral renal artery stenosis, the presence of fibrosis has been demonstrated in the normotensive non-hypertrophied right ventricle as well as the hypertensive, hypertrophied left ventricle. In contrast, infra renal aortic banding where RAAS is not activated did not result in cardiac fibrosis despite comparable increases in blood pressure and extent of LVH [31]. Supporting the argument for RAAS activation in the aetiolgy of myocardial fibrosis is the finding that it is not seen in LVH secondary to volume overload states such as atrial septal defects or arteriovenous fistulae where the pathway is not activated and therefore collagen concentration remains normal [32,33].
Angiotensin II stimulates the production of aldosterone and studies suggest both direct effects on the heart as well as indirect effects such as sodium retention, volume expansion and resultant hypervolaemia. Direct effects of aldosterone in the heart rely on the presence of mineralocorticoid receptors in cardiac cells. Silvestre, et al. demonstrated the existence of aldosterone synthase in the rat heart confirming synthesis adrenocortical hormones within the heart [34]. Subsequent studies have suggested the presence of a complete pathway resulting in aldosterone synthesis.
Brilla, et al. demonstrated that both aldosterone and angiotensin II could induce myocardial fibrosis in rats irrespective of LVH [35]. In vivo experiments have implied that aldosterone is involved in the production and deposition of collagen and therefore subsequent development of myocardial fibrosis. Uninephrectromised rats on a high sodium diet were exposed to 8 weeks of aldosterone infusion. CVF increased significantly at week 6 and week 8 in left and right ventricles with a progressive increase from week 4 [36]. Brilla and Weber demonstrated similar findings with uninephrectomised rats drinking 1% NaCL solution on an aldosterone infusion for 8 weeks. A significant increase in the deposition of interstitial and perivascular collagen was observed [37].
Patients with adrenal adenomas where there is chronic activation of RAAS demonstrate significant accumulation of collagen in the heart. Campbell, et al. evaluated autopsy proven adrenal adenomas and demonstrated the presence of reactive fibrosis in hypertrophied hearts [37]. Patients diagnosed with primary hyperaldosteronism who underwent adrenalectomy benefited from significant reductions in interventricular septal thickness, posterior wall thickness and LVM index [38]. In addition, pharmacological intervention with aldosterone antagonists such as spironolactone have revealed a reduction in circulating levels of procollagen type III N-terminal amino peptide in patients with stable chronic heart failure [39].
The Randomised Aldactone Evaluation Study (RALES) demonstrated a 30% reduction in mortality with the administration of spironolactone plus conventional therapy in patients with NYHA class III and IV symptomatic heart failure [40].
A study by Amanuma, et al. evaluated 29 patients with graded severities of hypertension (slight, moderate and severe) using endomyocardial biopsy of both the right and left ventricle. Breadth of the myocytes and the degree of interstitial fibrosis in both ventricles increased in proportion to the severity of hypertension [41]. These findings have also been observed in the interventricular septum [10] and the atria [42] supporting the argument for neurohormonal mechanisms in the development of fibrosis.
Consequences
Diastolic dysfunction
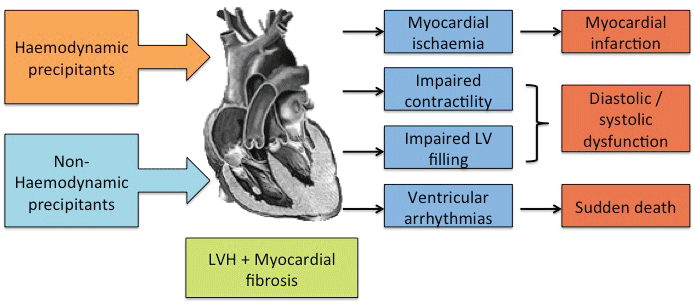
Diastole is comprised of an early phase, during which negative intraventricular pressure causes a suction effect and thus rapid filling. Diastasis is then followed by active filling, secondary to atrial contraction. In contrast to normal cardiac physiology where the majority of blood flow in diastole occurs during the early phase, in diastolic dysfunction there is reliance on atrial systole for as much as half of the filling volume. In patients with hypertensive heart disease and left ventricular hypertrophy, diastolic dysfunction is common (Figure 3).
.
Figure 3: Precipitants and clinical sequelae related to LVH and myocardial fibrosis. LVH = left ventricular hypertrophy, LV = left ventricle.
View Figure 3
Both animal and human studies have demonstrated that hypertension induced cardiac fibrosis results in diastolic dysfunction. Increased deposition of Type I collagen, which has a tensile strength comparable to steel, makes the increase in stiffness within myocardial tissue predictable. Diastolic heart failure accounts for up to 50% of heart failure in clinical practice and causes symptoms in a substantial number of hypertensive patients especially in the elderly [43].
Echocardiography is the modality of choice for assessing diastology and the use of tissue Doppler has demonstrated that even in hypertensive individuals without LVH, diastolic function is impaired [44]. The degree of myocardial fibrosis when assessed by collagen markers is inversely related to echocardiographic markers of diastolic function [3,4].
Previous rat models have been able to explicate the role of cytokines and inflammatory molecules such as TGF-β which acts via fibroblast activation. Kuwahara, et al. found that myocardial fibrosis could be prevented by blocking TGF-β activity thus improving diastolic dysfunction [45]. Further studies in rats have implicated macrophages in the development of myocardial fibrosis and it has been suggested that inhibiting inflammation using neutralising antibodies could attenuate the progression to diastolic dysfunction [46].
Systolic dysfunction
It is well documented that heart failure may occur in association with normal left ventricular ejection fraction (LVEF) or with reduced LVEF. Myocardial fibrosis, due to both ischaemic and non-ischaemic aetiologies is thought to play a pivotal role in the development and progression of systolic cardiac dysfunction [47] (Figure 3).
Conrad, et al. demonstrated that heart failure in the spontaneously hypertensive rat (SHR) was associated with histologically and biochemically determined myocardial fibrosis and impaired contractility when compared to non-failing SHR hearts and normotensive rats [25]. In humans, the greater the degree of myocardial fibrosis the greater the impairment of cardiac function, with advanced heart failure patients demonstrating more extensive myocardial fibrosis [48]. In end-stage heart failure a significant increase in CVF has been demonstrated.
There has long been an assumption that dilated cardiac failure occurs as a result of concentric LVH. This has previously been demonstrated in animal models as well as in humans with aortic stenosis and hypertrophic cardiomyopathy [49,50].
LVH may well be a precursor to LV systolic dysfunction independent of prior ischaemic events however myocardial infarction plays an important role in the progression to dilated heart failure in hypertensive patients. There is an association between LVH and subclinical atherosclerosis and a study using serial cardiac catheterization has also demonstrated that plaque rupture is more common in patients with LVH [51,52]. A retrospective study analysed 159 individuals with concentric LVH and normal LV function at baseline echocardiography. After an average follow-up period of four years a further study was performed demonstrating that only 18% had impaired LV function. Importantly almost half had suffered an MI in the interval period [53].
Arrhythmia and Sudden Death
Atrial fibrillation
In a study of almost 9000 hypertensive patients with no evidence of atrial fibrillation (AF) at baseline, echocardiographic evidence of LVH was a strong independent predictor of new onset AF [54]. This intimate relationship between hypertension and AF means that strict control of BP may prevent atrial fibrillation. The LIFE study also demonstrated that maintaining the systolic BP below 130 mmHg led to a reduced risk of new-onset AF compared to those with systolic BPs above 142 mmHg [55].
The prevention of diastolic dysfunction and atrial stretch using antihypertensives, in particular ACE inhibitors and ARBs, may also reduce the risk of AF. A meta-analysis of 56 308 patients found that RAS inhibitors reduced the relative risk of AF by 28% [56]. However there are mixed opinions, as only the LIFE study which recruited high- risk patients with LVH, demonstrated a benefit of losartan over atenolol in the reduction of AF. Studies recruiting from the general population did not demonstrate such a benefit. This may imply that prevention of AF is most effective in hypertensive patients with haemodynamic complications.
Ventricular arrhythmia
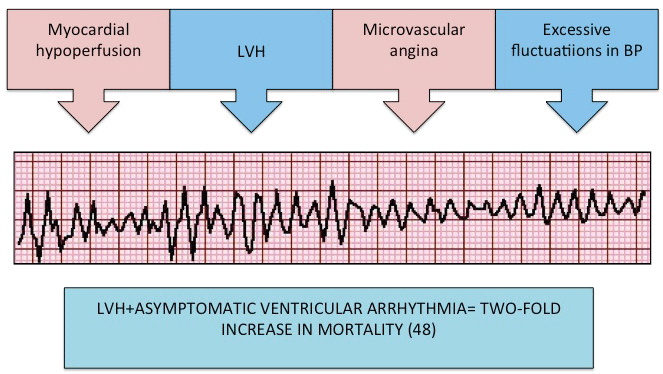
There is a significantly greater prevalence of ventricular arrhythmia and ventricular ectopy in patients with hypertension and LVH compared to those without LVH or normotensives [57] (Figure 3). The Framingham group has previously reported a two-fold increase in mortality in those with LVH and asymptomatic ventricular arrhythmias [58]. The extent of myocardial fibrosis present in the heart has been implicated in slowing of conduction and an increased susceptibility to arrhythmias due to interruption of coordinated transmission of electrical impulses. Other aetiological mechanisms include microvascular angina, which may occur in the absence of LVH, myocardial under perfusion due to disproportionate muscular hypertrophy compared to coronary artery growth and excess fluctuation in arterial pressure which may be arrhythmogenic [59-63] (Figure 4). LVH has thus been identified as an independent risk factor for the development of arrhythmia.
.
Figure 4: Aetiology of ventricular arrhythmias in hypertension. LVH = left ventricular hypertrophy, BP = blood pressure.
View Figure 4
Supporting this observation, a study in a cohort of 196 elderly hypertensive patients with LVH but without coronary artery disease demonstrated significant risk of developing ventricular fibrillation or sudden death compared to 358 patients without LVH (31% vs. 10%) [62].
Detection
Endomyocardial biopsies, although the gold-standard for assessment of myocardial CVF, have limited use in antemortem studies due to the invasive nature of the test. Other methods for evaluating cardiac fibrosis have been developed including echocardiographic parameters, measurement of hormones and biochemical markers [63-65].
Echocardiography
The use of ultrasound as an inexpensive, non-invasive assessment of myocardial tissue is well established using techniques such as M-mode and 2-dimensional evaluation of scar including akinesis and increased acoustic brightness. Tissue Doppler imaging may demonstrate reduced measures of cardiac function in the fibrotic heart, such as myocardial tissue velocity and deformation parameters. Assessment of longitudinal function in hypertensive hearts has been shown to be the most sensitive marker of subclinical disease. The mechanism reflects increased radial contraction to preserve cardiac function due to involvement of subendocardial and mid-wall fibres [66].
Echocardiographic deformation techniques i.e. strain and strain rates, are more sensitive than other echocardiographic techniques and may demonstrate abnormalities in early cardiomyopathic processes and can detect subclinical heart disease. The anti fibrotic effect of aldosterone antagonism has been demonstrated with improved parameters in diastolic dysfunction [67]. It must be noted that echocardiographic assessment of LV hypertrophy and geometry is limited by geometric assumptions which may result in reduced reproducibility, reliability and accuracy when compared to measurements made using CMR [68].
Cardiac magnetic resonance
CMR imaging is the non-invasive gold standard for the quantification of focal myocardial fibrosis. Myocardial fibrosis, which occurs secondary to ischaemia is easily detected using delayed contrast enhancement on cardiac magnetic resonance imaging (CMRI) [69]. However, this technique when used in hypertensive patients has previously demonstrated non-specific, non-ischaemic patterns of fibrosis [70]. A major flaw in utilising this method to assess diffuse myocardial fibrosis is that it is qualitative rather than quantitative. In addition, image contrast relies upon the difference in signal intensity between the areas of scarred myocardium and normal myocardium, but in non-ischaemic cardiomyopathy collagen deposition is typically diffuse resulting in a lack of regional scarring when evaluated with delayed contrast enhancement. A quantitative tissue characterisation method is available which measures the T1 relation time of the myocardium. The presence of expanded extracellular space secondary to myocardial fibrosis, scar formation or infiltration results in a greater distribution volume. The reduction in T1 relaxation time is thus more evident compared to normal myocardium. Using pre and post contrast T1 maps and the haematocrit, the extracellular volume fraction (ECV) may also be calculated. ECV has been shown to correlate with the extent of myocardial fibrosis and CVF [71].
Previous studies in hypertensive rat hearts and human papillary muscles have identified variation of the T1 mapping time with fibrosis, which correlates with myocardial collagen composition [72,73].
A recent study by Treibel, et al. demonstrated diffuse myocardial fibrosis evaluated by T1 mapping in hypertensive patients was raised but only occurred in those with LVH [74]. This study implies that hypertensive patients exhibit both diffuse and focal myocardial fibrosis associated with LVH. Kuruvilla, et al. documented that hypertensive patients with LVH exhibited higher extracellular volumes and longer native T1 mapping times with an associated decrease in peak systolic circumferential strain in contrast to hypertensive patients with no LVH and controls [75]. CMR T1 mapping has great potential for evaluating cardiac fibrosis in hypertensive patients.
Treatment and Regression of Myocardial Fibrosis
Regression of myocardial fibrosis with pharmacological therapy is an important prognostic intervention for patients with hypertension. Various studies have indicated functional benefits, particularly with the use of ACE Inhibitors and angiotensin receptor blockers (ARBs) [76-78] .
Animal studies evaluating the efficacy of antihypertensive drugs in myocardial regression demonstrated zofenapril and labetalol could regress myocardial fibrosis in rats with renovascular hypertension and LVH [79].
Specific to diastolic dysfunction Brilla, et al. examined male spontaneously hypertensive rats (SHRs) with cardiac fibrosis and LVH, and administered a 12 week course of ACE inhibitor. The high dose normalised arterial blood pressure and regressed LVH, the low dose achieved neither end point. Rats in both high and low dose treatment groups demonstrated regression in cardiac fibrosis as well as recovery of diastolic function to a level comparable with normotensive control rats [Wistar-Kyoto rats (WKYs)] [80]. Importantly these findings emphasise the disassociation between LVH and left ventricular stiffness, and the significance of myocardial quality over quantity. A further SHR study by the same group evaluated older rats with more established fibrosis and LVH. They were treated with a longer course of lisinopril, which resulted in partial regression of fibrosis and myocardial stiffness to measurements comparable to age-matched WKY controls [81]. Subsequently Brilla and colleagues published a pivotal study evaluating the effects of lisinopril versus hydrochlorothiazide in patients with established hypertension, LVH and diastolic dysfunction on echocardiography [82]. Endomyocardial biopsy was performed before and after treatment and morphological (CVF) and biochemical (hydroxyproline) parameters were used to quantify fibrosis. Regression of myocardial fibrosis and improvement in echocardiographic measurements of diastolic stiffness were demonstrated in the lisinopril group after six months of therapy.
In a study by DIez, et al. hypertensive patients with severe fibrosis assessed using endomyocardial biopsy demonstrated significant reductions in CVF and left ventricular chamber stiffness after 12 months of treatment with losartan [83]. The Losartan Intervention for Endpoint Reduction in Hypertension (LIFE) study demonstrated a reduction of LVH in response to losartan, independent of lowering blood pressure [84].
Interference with collagen turnover using ACE-inhibitors and angiotensin receptor blockers appears to have greater effect on diastolic dysfunction than reduction in arterial pressure or regression of LVH.
Attenuation of ventricular arrhythmias can also be achieved by reducing LVH in hypertensive patients. A reduction in ventricular ectopy has been demonstrated with the use of calcium channel blockers, β-blockers and ACE inhibitors suggesting the anti-arrhythmic effect is associated with the reduction in LVH rather than the effects of the drug only. Gonzalez-Fernandez, et al. demonstrated a significant reduction in LVH and associated ventricular ectopy with the use of an ACE inhibitor compared to progression of LVH and no change in the frequency of ventricular ectopy in the placebo group [85].
The Aliskiren in Left Ventricular Hypertrophy (ALLAY) study demonstrated that Aliskiren (acting to inhibit renin) alone or in combination with losartan was associated with a greater reduction in aldosterone compared to losartan alone. The reduction in plasma aldosterone was associated with a corresponding regression of LV wall thickness and LV myocardial index reduction. This study provides supporting evidence that regression of LVH may be caused by aldosterone inhibition [77].
A meta-analysis of 39 clinical trials found that ACE Inhibitors were the most potent at reducing left ventricular mass index. Calcium channel blockers had an intermediate effect with beta blockers and diuretics having even less effect [86]. A study supporting this showed that telmisartan reduced LV mass to greater extent than hydrochlorothiazide [87]. The same group demonstrated a similar finding when comparing telmisartan with carvedilol [88]. Similar studies using CMR to assess changes in LVH and LV mass support this finding. Due to the widespread availability and safety of echocardiography, it is the cornerstone of cardiac imaging in cardiomyopathy. However, a study of more than 200 hypertensives demonstrated that Eplerenone wasjust as effective compared to enalapril in both LVH regression and blood pressure control. LV mass and systolic blood pressure could be reduced more effectively by combining eplerenone and enalapril [89]. It is likely that more studies will arise using CMR to assess LV geometry and the effects of pharmacological intervention on these parameters.
The importance of identifying patients with hypertension and LVH cannot be underestimated when there is such convincing evidence that anti-hypertensive drugs have the potential to reduce morbidity and mortality in these individuals.
Prognosis
Despite the initial adaptive response to systemic hypertension, LVH is associated with a two-fold increase in cardiovascular morbidity and mortality. Studies suggest that intervention resulting regression of LVH and cardiac fibrosis may improve survival in hypertensive individuals. This survival benefit may be independent of blood pressure reduction with the use of certain pharmacological treatments [76]. In a study of 151 hypertensive patients followed up after 10 years with serial echocardiography, lack of LVH regression was associated with higher risk of cardiovascular events [90]. Moreover this risk was almost normalised in those who demonstrated LVH regression.
It follows then that specific interventions resulting in regression of LVH and cardiac fibrosis should be the mainstay of treatment to prevent cardiovascular complications. Pursuit of lower blood pressure readings may also be inconsequential since pharmacological mechanisms appear to act independently.
Conclusion
Cardiac fibrosis and LVH secondary to hypertensive heart disease has been identified as a significant cause of morbidity and mortality. Hypertension remains one of the top 15 causes of global mortality and is predicted to become more prevalent over the coming decade. Deemed a 'silent killer' due to the lack of symptoms and the lethal nature of cardiovascular and cerebrovascular complications, prevention and treatment remain challenging for the physician. However, over recent years progress has been made in the understanding of underlying mechanisms by which myocardial architecture is disrupted by the hypertensive disease process. Our aim should be to harness this knowledge to prevent cardiac fibrosis in the first instance, or at least to regress disease with pharmacological intervention in order to improve outcomes for our hypertensive patients.
References
-
Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, et al. (2005) Global burden of hypertension: analysis of worldwide data. Lancet 365: 217-223.
-
World Health Organization (2013) A global brief on Hyper tension World Health Day 2013. World Health Organization.
-
Müller-Brunotte R, Kahan T, Lopez B, Edner M, González A, et al. (2007) Myocardial fibrosis and diastolic dysfunction in patients with hypertension: results from the Swedish Irbesartan Left Ventricular Hypertrophy Investigation versus Atenolol (SILVHIA). J Hypertens 25: 1958-1966.
-
Martos R, Baugh J, Ledwidge M, O'Loughlin C, Conlon C, et al. (2007) Diastolic heart failure: Evidence of increased myocardial collagen turnover linked to diastolic dysfunction. Circulation 115: 888-895.
-
Laviades C, Varo N, Fernandez J, Mayor G, Gil MJ, et al. (1998 ) Abnormalities of the Extracellular Degradation of Collagen Type I in Essential Hypertension. Circulation 98: 535-540.
-
Weber KT, Sun Y, Guarda E, Katwa LC, Ratajska A, et al. (1995) Myocardial fibrosis in hypertensive heart disease: an overview of potential regulatory mechanisms. Eur Hear J 16 Suppl C: 24-28.
-
Kai H, Kuwahara F, Tokuda K, Imaizumi T (2005) Diastolic dysfunction in hypertensive hearts: roles of perivascular inflammation and reactive myocardial fibrosis. Hypertens Res 28: 483-490.
-
López B, González A, Querejeta R, Díez J (2005) The use of collagen-derived serum peptides for the clinical assessment of hypertensive heart disease. J Hypertens 23: 1445-1451.
-
Diez J, Laviades C, Mayor G, Gil MJ, Monreal I (1995) Increased Serum Concentrations of Procollagen Peptides in Essential Hypertension: Relation to Cardiac Alterations. Circulation 91: 1450-1456.
-
Querejeta R, Varo N, López B, Larman M, Artiñano E, et al. (2000) Serum carboxy-terminal propeptide of procollagen type I is a marker of myocardial fibrosis in hypertensive heart disease. Circulation 101: 1729-1735.
-
López B, Querejeta R, Varo N, González a, Larman M, et al. (2001) Usefulness of serum carboxy-terminal propeptide of procollagen type I in assessment of the cardioreparative ability of antihypertensive treatment in hypertensive patients. Circulation 104: 286-291.
-
Lindsay MM, Maxwell P, Dunn FG (2002) TIMP-1: A marker of left ventricular diastolic dysfunction and fibrosis in hypertension. Hypertension 40: 136-141.
-
Varo N, Iraburu MJ, Varela M, López B, Etayo JC, et al. ( 2000) Chronic AT(1) blockade stimulates extracellular collagen type I degradation and reverses myocardial fibrosis in spontaneously hypertensive rats. Hypertension 35: 1197-1202.
-
Varo N, Etayo JC, Zalba G, Beaumont J, Iraburu MJ, et al. (1999) Losartan inhibits the post-transcriptional synthesis of collagen type I and reverses left ventricular fibrosis in spontaneously hypertensive rats. J Hypertens 17: 107-114.
-
Bishop JE, Lindahl G (1999) Regulation of cardiovascular collagen synthesis by mechanical load. Cardiovasc Res 42: 27-44.
-
Lorell BH, Carabello BA (2000) Left ventricular hypertrophy: pathogenesis, detection, and prognosis. Circulation 102: 470-479.
-
Zaidi A, Sharma S (2013) Exercise and heart disease: from athletes and arrhythmias to hypertrophic cardiomyopathy and congenital heart disease. Future Cardiol 9: 119-136.
-
Levy D, Larson MG, Vasan RS, Kannel WB, Ho KK (1996) The Progression From Hypertension to Congestive Heart Failure. JAMA 275: 1557-1562.
-
Verdecchia P, Angeli F, Borgioni C, Gattobigio R, de Simone G, et al. (2003) Changes in cardiovascular risk by reduction of left ventricular mass in hypertension: a meta-analysis. Am J Hypertens 16: 895-899.
-
Fagard RH, Celis H, Thijs L, Wouters S (2009) Regression of left ventricular mass by antihypertensive treatment: A meta-analysis of randomized comparative studies. Hypertension 54: 1084-1091.
-
Drazner MH, Dries DL, Peshock RM, Cooper RS, Klassen C, et al. (2005) Left ventricular hypertrophy is more prevalent in blacks than whites in the general population: The Dallas heart study. Hypertension. 46: 124-129.
-
Sharp A, Tapp R, Francis DP, McG Thom SA, Hughes AD, et al. (2008) Ethnicity and Left Ventricular Diastolic Function in Hypertension. An ASCOT (Anglo-Scandinavian Cardiac Outcomes Trial) Substudy. J Am Coll Cardiol 52: 1015-1021.
-
Frohlich ED, Apstein C, Chobanian AV, Devereux RB, Dustan HP, et al. (1992) The Heart in Hypertension. N Engl J Med 327: 998-1008.
-
Drazner MH, Rame JE, Marino EK, Gottdiener JS, Kitzman DW, et al. (2004) Increased left ventricular mass is a risk factor for the development of a depressed left ventricular ejection fraction within five years: The Cardiovascular Health Study. J Am Coll Cardiol 43: 2207-2215.
-
Conrad CH, Brooks WW, Hayes JA, Sen S, Robinson KG, et al. (1995) Myocardial fibrosis and stiffness with hypertrophy and heart failure in the spontaneously hypertensive rat. Circulation 91: 161-170.
-
Verdecchia P, Staessen JA, Angeli F, de Simone G, Achilli A, et al. (2009) Usual versus tight control of systolic blood pressure in non-diabetic patients with hypertension (Cardio-Sis): an open-label randomised trial. Lancet 374: 525-533.
-
Rossi MA (1998) Pathologic fibrosis and connective tissue matrix in left ventricular hypertrophy due to chronic arterial hypertension in humans. J Hypertens 16: 1031-1041.
-
Tanaka M, Fujiwara H, Onodera T, Wu DJ, Hamashima Y, et al. (1986) Quantitative analysis of myocardial fibrosis in normals, hypertensive hearts, and hypertrophic cardiomyopathy. Br Heart J 55: 575-581.
-
Reddy HK, Campbell SE, Janicki JS, Zhou G, Weber KT (1993) Coronary microvascular fluid flux and permeability: influence of angiotensin II, aldosterone, and acute arterial hypertension. J Lab Clin Med 121: 510-521.
-
Hao J, Wang B, Jones SC, Jassal DS, Dixon IM (2000) Interaction between angiotensin II and Smad proteins in fibroblasts in failing heart and in vitro. Am J Physiol Heart Circ Physiol 279: H3020-H3030.
-
Brilla CG, Pick R, Tan LB, Janicki JS, Weber KT (1990) Remodeling of the Rat Right and Left Ventricles in Experimental Hypertension. Circ Res 67: 1355-1364.
-
Marino TA, Kent RL, Uboh CE, Fernandez E, Thompson EW, et al. (1985) Structural analysis of pressure versus volume overload hypertrophy of cat right ventricle. Am J Physiol 249: H371-H379.
-
Weber KT, Brilla CG (1991) Pathological hypertrophy and cardiac interstitium. Fibrosis and renin-angiotensin-aldosterone system. Circulation 83: 1849-1865.
-
Silvestre JS, Robert V, Heymes C, Aupetit-Faisant B, Mouas C, et al. (1998) Myocardial production of aldosterone and corticosterone in the rat: Physiological regulation. J Biol Chem 273: 4883-4891.
-
Brilla CG, Zhou G, Matsubara L, Weber KT (1994) Collagen metabolism in cultured adult rat cardiac fibroblasts: response to angiotensin II and aldosterone. J Mol Cell Cardiol 26: 809-820.
-
Campbell SE, Janicki JS, Weber KT (1995) Temporal differences in fibroblast proliferation and phenotype expression in response to chronic administration of angiotensin II or aldosterone. J Mol Cell Cardiol 27: 1545-1560.
-
Campbell SE, Diaz-Arias AA, Weber KT (1992) Fibrosis of the human heart and systemic organs in adrenal adenoma. Blood Press 1: 149-156.
-
Denolle T, Chatellier G, Julien J, Battaglia C, Luo P, et al. (1993) Left ventricular mass and geometry before and after etiologic treatment in renovascular hypertension, aldosterone-producing adenoma, and pheochromocytoma. Am J Hypertens 6: 907-913.
-
MacFadyen RJ, Barr CS, Struthers AD (1997) Aldosterone blockade reduces vascular collagen turnover, improves heart rate variability and reduces early morning rise in heart rate in heart failure patients. Cardiovasc Res 35: 30-34.
-
Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, et al. (1999) The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 341: 709-717.
-
Amanuma S, Sekiguchi M, Ogasawara S, Honda M, Hosoda S (1994) Biventricular endomyocardial biopsy findings in essential hypertension of graded severity. Postgrad Med J: S67-S71.
-
A Boldt, U Wetzel, J Lauschke, J Weigl, J Gummert, et al. (2004) Fibrosis in left atrial tissue of patients with atrial fibrillation with and without underlying mitral valve disease. Heart 90: 400-405.
-
Vasan RS, Larson MG, Benjamin EJ, Evans JC, Reiss CK, et al. (1999) Congestive heart failure in subjects with normal versus reduced left ventricular ejection fraction: prevalence and mortality in a population-based cohort. J Am Coll Cardiol 33: 1948-1955.
-
Muller Brunotte R, Kahan T, Malmqvist K, Ring M, Edner M (2006 ) Tissue Velocity Echocardiography Shows Early Improvement in Diastolic Function With Irbesartan and Atenolol Therapy in Patients With Hypertensive Left Ventricular Hypertrophy: Results From the Swedish Irbesartan Left Ventricular Hypertrophy Investigation. Am J Hypertens 19: 927-936.
-
Kuwahara F, Kai H, Tokuda K, Kai M, Takeshita A, et al. (2002) Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation. 106: 130-135.
-
Kuwahara F, Kai H, Tokuda K, Takeya M, Takeshita A, et al. (2004) Hypertensive Myocardial Fibrosis and Diastolic Dysfunction: Another Model of Inflammation?. Hypertension 43: 739-745.
-
Mann DL (1999) Mechanisms and Models in Heart Failure: A Combinatorial Approach . Circ 100: 999-1008.
-
Sun Y, Weber KT (1998) Cardiac remodelling by fibrous tissue: role of local factors and circulating hormones. Ann Med 1: 3-8.
-
Rapaport E (1975) Natural history of aortic and mitral valve disease. Am J Cardiol 35: 221-227.
-
Spirito P, Maron BJ, Bonow RO, Epstein SE (1987) Occurrence and significance of progressive left ventricular wall thinning and relative cavity dilatation in hypertrophic cardiomyopathy. Am J Cardiol 60: 123-129.
-
Mehta SK, Rame JE, Khera A, Murphy SA, Canham RM, et al. (2007) Left ventricular hypertrophy, subclinical atherosclerosis, and inflammation. Hypertension 49: 1385-1391.
-
Heidland UE, Strauer BE (2001) Left ventricular muscle mass and elevated heart rate are associated with coronary plaque disruption. Circulation 104: 1477-1482.
-
Rame JE, Ramilo M, Spencer N, Blewett C, Mehta SK, et al. (2004) Development of a depressed left ventricular ejection fraction in patients with left ventricular hypertrophy and a normal ejection fraction. Am J Cardiol 93: 234-237.
-
Wachtell K, Hornestam B, Lehto M, Slotwiner DJ, Gerdts E, et al. (2005) Cardiovascular morbidity and mortality in hypertensive patients with a history of atrial fibrillation: The Losartan Intervention for End point reduction in hypertension (LIFE) study. J Am Coll Cardiol 45: 705-711.
-
Okin PM, Hille DA, Larstorp AC, Wachtell K, Kjeldsen SE, et al. (2015) Effect of Lower On-Treatment Systolic Blood Pressure on the Risk of Atrial Fibrillation in Hypertensive Patients. Hypertension. 66: 368-373.
-
Healey JS, Baranchuk A, Crystal E, Morillo CA, Garfinkle M, et al. (2005) Prevention of atrial fibrillation with angiotensin-converting enzyme inhibitors and angiotensin receptor blockers: A meta-analysis. J Am Coll Cardiol 45: 1832-1839.
-
Messerli FH, Ventura HO, Elizardi DJ, Dunn FG, Frohlich ED (1984) Hypertension and sudden death. Increased ventricular ectopic activity in left ventricular hypertrophy. Am J Med 77: 18-22.
-
Bikkina M, Larson MG, Levy D (1993) Asymptomatic ventricular arrhythmias and mortality risk in subjects with left ventricular hypertrophy. J Am Coll Cardiol 22: 1111-1116.
-
Sideris DA (1993) High blood pressure and ventricular arrhythmias. Eur Heart J 14: 1548-1553.
-
Houghton JL, Prisant LM, Carr AA, von Dohlen TW, Frank MJ (1991) Relationship of left ventricular mass to impairment of coronary vasodilator reserve in hypertensive heart disease. Am Heart J 121: 1107-1112.
-
Harrison DG, Marcus ML, Dellsperger KC, Lamping KG, Tomanek RJ (1991) Pathophysiology of myocardial perfusion in hypertension. Circulation 83: 14-18.
-
Aronow WS, Epstein S, Koenigsberg M, Schwartz KS (1988) Usefulness of echocardiographic left ventricular hypertrophy, ventricular tachycardia and complex ventricular arrhythmias in predicting ventricular fibrillation or sudden cardiac death in elderly patients. Am J Cardiol 62: 1124-1125.
-
Mitchell JA, Ventura HO, Mehra MR (2005) Early recognition and treatment of hypertensive heart disease. Curr Opin Cardiol 20: 282-289.
-
Collier P, Watson CJ, Voon V, Phelan D, Jan A, et al. (2011) Can emerging biomarkers of myocardial remodelling identify asymptomatic hypertensive patients at risk for diastolic dysfunction and diastolic heart failure? Eur J Heart Fail 13: 1087-1095.
-
Janardhanan R, Kramer CM (2011) Imaging in hypertensive heart disease. Expert Rev Cardiovasc Ther 9: 199-209.
-
Fang ZY, Leano R, Marwick TH (2004) Relationship between longitudinal and radial contractility in subclinical diabetic heart disease. Clin Sci (Lond) 106: 53-60.
-
Mottram PM, Haluska B, Leano R, Cowley D, Stowasser M, et al. (2004) Effect of aldosterone antagonism on myocardial dysfunction in hypertensive patients with diastolic heart failure. Circulation 110: 558-565.
-
Bottini PB, Carr AA, Prisant LM, Flickinger FW, Allison JD, et al. (1995) Magnetic resonance imaging compared to echocardiography to assess left ventricular mass in the hypertensive patient. Am J Hypertens 8: 221-228.
-
Kim RJ, Wu E, Rafael A, Chen EL, Parker MA, et al. (2000) The Use of Contrast-Enhanced Magnetic Resonance Imaging to Identify Reversible Myocardial Dysfunction. N Engl J Med 343: 1445-1453.
-
Rudolph A, Abdel-Aty H, Bohl S, Boyé P, Zagrosek A, et al. (2009) Noninvasive Detection of Fibrosis Applying Contrast-Enhanced Cardiac Magnetic Resonance in Different Forms of Left Ventricular Hypertrophy. Relation to Remodeling. J Am Coll Cardiol 53: 284-291.
-
de Meester de Ravenstein C, Bouzin C, Lazam S, Boulif J, Amzulescu M, et al. (2015) Histological Validation of measurement of diffuse interstitial myocardial fibrosis by myocardial extravascular volume fraction from Modified Look-Locker imaging (MOLLI) T1 mapping at 3 T. J Cardiovasc Magn Reson 17: 48.
-
Toni R, Boicelli CA, Baldassarri AM (1986) Characterization of human pathological papillary muscles by 1H-NMR spectroscopic and histologic analysis. Int J Cardiol 11: 231-234.
-
Grover-McKay M, Scholz TD, Burns TL, Skorton DJ (1991) Myocardial collagen concentration and nuclear magnetic resonance relaxation times in the spontaneously hypertensive rat. Invest Radiol 26: 227-232.
-
Treibel TA, Zemrak F, Sado DM, Banypersad SM, White SK, et al. (2015) Extracellular volume quantification in isolated hypertension - changes at the detectable limits?. J Cardiovasc Magn Reson 17: 74.
-
Kuruvilla S, Janardhanan R, Antkowiak P, Keeley EC, Adenaw N, et al. (2015) Increased Extracellular Volume and Altered Mechanics Are Associated With LVH in Hypertensive Heart Disease, Not Hypertension Alone. JACC Cardiovasc Imaging 8: 172-180.
-
Brilla C, Funck R, Rupp H (2000) Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circulation 102: 1388-1393.
-
Pouleur AC, Uno H, Prescott MF, Desai A, Appelbaum E, et al. (2011) Suppression of aldosterone mediates regression of left ventricular hypertrophy in patients with hypertension. J Renin-Angiotensin-Aldosterone Syst 12: 483-490.
-
Bos R, Mougenot N, Findji L, Mediani O, Vanhoutte PM, et al. (2005) Inhibition of catecholamine-induced cardiac fibrosis by an aldosterone antagonist. J Cardiovasc Pharmacol 45: 8-13.
-
Brilla CG (2000) Regression of myocardial fibrosis in hypertensive heart disease: diverse effects of various antihypertensive drugs. Cardiovasc Res 46: 324-331.
-
Brilla CG, Janicki JS, Weber KT (1991) Impaired diastolic function and coronary reserve in genetic hypertension. Role of interstitial fibrosis and medial thickening of intramyocardial coronary arteries. Circ Res 69: 107-115.
-
Brilla CG, Matsubara L, Weber KT (1996) Advanced hypertensive heart disease in spontaneously hypertensive rats. Lisinopril-mediated regression of myocardial fibrosis. Hypertension 28 269-275.
-
Brilla CG, Funck RC, Rupp H (2000) Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circulation 102: 1388-1393.
-
Díez J, Querejeta R, López B, González A, Larman M, et al. (2002) Losartan-dependent regression of myocardial fibrosis is associated with reduction of left ventricular chamber stiffness in hypertensive patients. Circulation 105: 2512-2517.
-
Okin PM, Devereux RB, Jern S, Kjeldsen SE, Julius S, et al. (2004) Regression of electrocardiographic left ventricular hypertrophy during antihypertensive treatment and the prediction of major cardiovascular events. JAMA 292: 2343-2349.
-
Gonzalez-Fernandez RA, Rivera M, Rodriguez PJ, Fernandez-Martinez J, Soltero LH, et al. (1993) Prevalence of ectopic ventricular activity after left ventricular mass regression. Am J Hypertens 6: 308-13.
-
Schmieder RE, Martus P, Klingbeil A (1996) Reversal of left ventricular hypertrophy in essential hypertension. A meta-analysis of randomized double-blind studies. JAMA 275: 1507-1513.
-
Galzerano D, Tammaro P, Cerciello A, Breglio R, Mallardo M, et al. (2004) Freehand three-dimensional echocardiographic evaluation of the effect of telmisartan compared with hydrochlorothiazide on left ventricular mass in hypertensive patients with mild-to-moderate hypertension: a multicentre study. J Hum Hypertens 18: 53-59.
-
Galzerano D, Tammaro P, Del Viscovo L, Lama D, Galzerano A, et al. (2005) Three-dimensional echocardiographic and magnetic resonance assessment of the effect of telmisartan compared with carvedilol on left ventricular mass: A multicenter, randomized, longitudinal study. Am J Hypertens 18: 1563-1569.
-
Pitt B, Reichek N, Willenbrock R, Zannad F, Phillips RA, et al. (2003) Effects of Eplerenone, Enalapril, and Eplerenone/Enalapril in Patients With Essential Hypertension and Left Ventricular Hypertrophy: The 4E-Left Ventricular Hypertrophy Study. Circulation 108: 1831-1838.
-
Muiesan ML, Salvetti M, Rizzoni D, Castellano M, Donato F, et al. (1995) Association of change in left ventricular mass with prognosis during long-term antihypertensive treatment. J Hypertens 13: 1091-1095.
