

Obstetrics and Gynaecology Cases - Reviews
Comparative Genomic Hybridization (CGH) for Prenatal Diagnosis of Wolf-Hirschhorn Syndrome
Adara Benitez Martin1*, Marina Naveiro Fuentes1, Manuel Vargas Perez2, Maria Setefilla Lopez Criado1, Carmen Entrala Bernal1 and Sebastian Manzanares Galan1
1Departament of Obstetrics and Gynecology, Virgen de las Nieves University Hospital, Spain
2Departament of Pediatrics and Specific Areas, San Cecilio University Hospital, Spain
*Corresponding author: Adara Benitez Martin, Departament of Obstetrics and Gynecology, Virgen de las Nieves University Hospital, Granada, Spain, Tel: 34-654-204-006; E-mail: adarabm@gmail.com
Obstet Gynecol Cases Rev, OGCR-1-009, (Volume 1, Issue 2), Original Article; ISSN: 2377-9004
Received: October 07, 2014 | Accepted: November 14, 2014 | Published: November 17, 2014
Citation: Benitez A, Naveiro M, Vargas M, López-Criado MS, Entrala C, et al. (2014) Comparative Genomic Hybridization (CGH) for Prenatal Diagnosis of Wolf-Hirschhorn Syndrome. Gynecol Cases Rev 1:009. 10.23937/2377-9004/1410009
Copyright: © 2014 Martin AB, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Wolf-Hirschhorn Syndrome (WHS) is a rare genetic condition caused by partial deletion of the short arm of chromosome 4 (4p-). Variability in 4p deletions and rearrangements leads to a wide spectrum of clinical manifestations of this disease. Most prenatal WHS diagnoses are associated with large 4p deletions identified by conventional cytogenetic techniques; however some submicroscopic deletions can only be diagnosed using molecular techniques. In this case report, a combined diagnostic approach based on conventional karyotyping and comparative genomic hybridization (CGH) offered a quick and definitive result to allow accurate prognoses and genetic counseling of WHS for the family.
Keywords
Wolf-hirschhorn syndrome; Array-comparative genomic hybridization (array-CGH); Prenatal diagnosis; 4p16.3 deletion; "Greek warrior" helmet profile.
Background
Wolf-Hirschhorn Syndrome (WHS), also known as deletion 4p or 4p- syndrome, is a well known clinical condition caused by deletions of variable amplitude in the chromosomal region 4p16.3 [1]. WHS was first described in 1965 in two independent publications by Wolf et al. [2] and Hirschhorn et al. [3]. The prevalence of this syndrome varies between 1:20.000 and 1:50.000 births, with a 2:1 bias in favor of females [4].
Factors for the great phenotypic variability that characterizes WHS include extent of the 4p deletion, the complexity of the basic genomic defect and the severity of seizures. However, the main clinical features include: pre and postnatal growth delay, profound psychomotor retardation, seizures, skeletal abnormalities, craniofacial dysgenesis (microcephaly, prominent glabella, widely spaced and prominent eyes, a "Greek warrior helmet appearance" of the nose, hypertelorism, cleft lip/palate, plump lips), heart defects and urinary tract malformations [5-9].
Most prenatally diagnosed cases of WHS are associated with large 4p deletions identified by conventional karyotype. However, with the widespread use of new molecular techniques such as array comparative genomic hybridization (a-CGH), the diagnosis of submicroscopic chromosomal aberrations associated with WHS critical regions could be improved, as is currently the case for many other complex genetic syndromes [5].
Clinical Report
The patient was a 31-year-old healthy primigravida with no medical or surgical history, no Diabetes Mellitus, no toxic habits, no malformations or genetic syndromes in her family or that of her husband, and non-consanguineous parents. The patient received regular treatments with multivitamins including potassium iodine and folic acid. The first trimester ultrasound, performed with an abdominal probe (Voluson ProV), showed a live fetus with crown-rump length of 66.7mm, equivalent to 13 weeks and 2 days gestation, and a nuchal translucency of 1.5 mm. Additional chromosomal markers were normal (nasal bone present and positive a-wave flow in the Dutus Venosus), except for the presence of tricuspid regurgitation with normal cardiac morphology. No other structural malformations were observed.
Biochemical markers in the first trimester, analyzed by Elecsys analyzer (Roche�), were in the normal range (B-hCG: 0.319 MoMs, PAPP-A: 1.828 MoMs). The risk for chromosomal abnormalities was calculated by using the FMF� module of the Astraia computer system, and was reported to be 1:3.833 for trisomy 21 and 1:20.000 for trisomy 13 and 18.
In the 20th week, a routine ultrasound, performed with an abdominal probe and ultrasound machine Toshiba Xsario X6, revealed a live fetus with biometry according to symmetric growth restriction (3rd centile for the gestational age) and estimated fetal weight, by Hadlock algorythm, of 201 grams. The ultrasound evaluation also showed a hypoplastic nasal bone (1.9mm) (Figure 1) without nuchal edema collapsed stomach and a single umbilical artery (Figure 2). Tricuspid regurgitation persisted and the fetal heart ultrasound was once again normal (Figure 3). Because of the risk of a genetic condition in the fetus, an amniotic fluid study was offered to the patient, and amniocentesis was carried out.
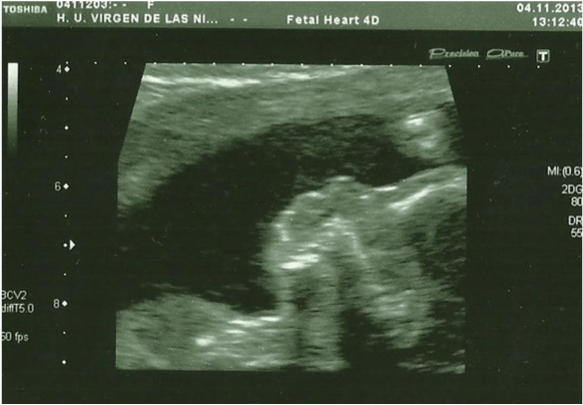 Figure 1: In the 20th week, the ultrasound evaluation show a hypoplastic
nasal bone, measuring 1,9mm (abdominal probe, Toshiba Xsario X6).
View Figure 1
Figure 1: In the 20th week, the ultrasound evaluation show a hypoplastic
nasal bone, measuring 1,9mm (abdominal probe, Toshiba Xsario X6).
View Figure 1
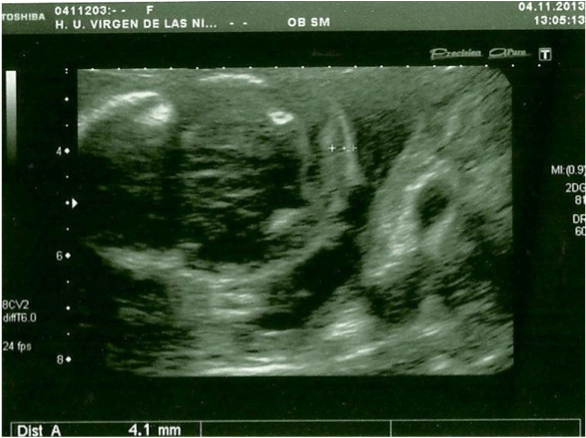 Figure 2: Axial prenatal ultrasonogram of a fetal head reveal normal nuchal
translucency (abdominal probe, Toshiba Xsario X6).
View Figure 2
Figure 2: Axial prenatal ultrasonogram of a fetal head reveal normal nuchal
translucency (abdominal probe, Toshiba Xsario X6).
View Figure 2
 Figure 3: Fetal heart ultrasound in the 20th week, Tricuspid regurgitation.
View Figure 3
Figure 3: Fetal heart ultrasound in the 20th week, Tricuspid regurgitation.
View Figure 3
qF-PCR, conventional karyotyping and array-CGH were requested. 7 days after the procedure the results of qF-PCR reported normal 21, 13, 18 and sex chromosomes. 14 days after the procedure, array-CGH indicated a pathogenic duplication in the 3p26.3p24.3 cytoband of 19,7 Megabases (Mb) and 78 genes OMIM, as well as pathogenic deletion in the 4p16.3p16.1 cytoband of 9.5 Mb and 67 genes OMIM, affecting the entire WHS critical region and suggesting an unbalanced rearrangement between chromosomes 3 and 4 [t (3, 4) (p24.3, p16, 1)]. A month after the amniocentesis, karyotype information showed an addition on the short arm of chromosome 4 with an atypical pattern of bands that could not be determined (Figure 4). Based on these results, prenatal diagnosis of WHS was made and the patient decided to terminate pregnancy at 24 weeks gestation. A female fetus weighing 300 grams was obtained. Pathologic examination showed no significant phenotypic or morphological alterations.
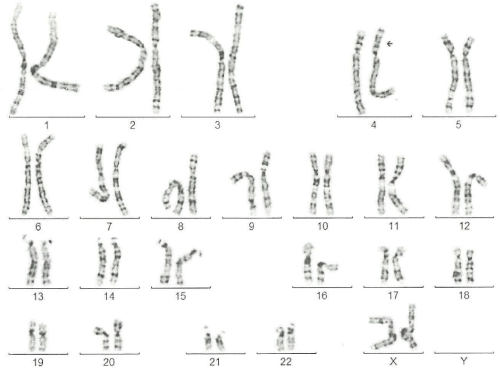 Figure 4: a-CGH profile of chromosome 4 showing a terminal deletion. To
the right, the whole chromosomes 4 view. To the left, the enlarged view of
the rearrangement as provided by Labco. The overall size of the deletion
was about 9.5Mb.
View Figure 4
Figure 4: a-CGH profile of chromosome 4 showing a terminal deletion. To
the right, the whole chromosomes 4 view. To the left, the enlarged view of
the rearrangement as provided by Labco. The overall size of the deletion
was about 9.5Mb.
View Figure 4
The mother�s karyotype was normal (46, XX), while in the father�s has been detected the translocation between the short arm of chromosome 3 and chromosome 4, with karyotype: 46, XY t (3,4)(p24.1;p16.1) (Figure 5).
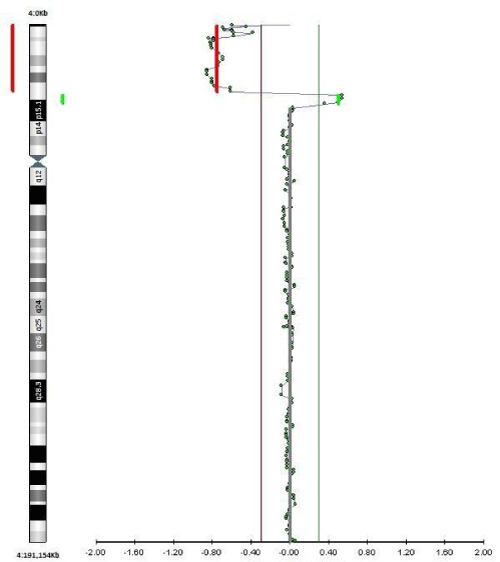 Figure 5: A karyotype 46 XX, Addition on the short arm of chromosome 4
with an atypical pattern of bands that could not be determined A karyotype
46 XX, Addition on the short arm of chromosome 4 with an atypical pattern
of bands that could not be determined.
View Figure 5
Figure 5: A karyotype 46 XX, Addition on the short arm of chromosome 4
with an atypical pattern of bands that could not be determined A karyotype
46 XX, Addition on the short arm of chromosome 4 with an atypical pattern
of bands that could not be determined.
View Figure 5
Discussion
WHS is a well-known genetic disease caused by partial deletion of the short arm of chromosome 4. Clinical manifestations in WHS vary greatly in individual patients, depending on the variability in the extent of the 4p deletion, and on the complexity of the basic genomic defect, which includes not only isolated deletion, but also double chromosome imbalances as a consequence of unbalanced translocations, unbalanced inversions and dup/del 4p.
In prenatal diagnosis of WHS, the main sonographic finding is a severe and early intrauterine growth restriction (percentile< 3) which may be associated with multiple congenital malformations such as craniofacial abnormalities (microcephaly, hypertelorism, prominent glabella, high forehead and low-set ears), midline defects like corpus callosum agenesis, cleft lip and palate, and atrial or ventricular septal defects [7,10]. Other malformations such as cystic hygroma, ventricular cysts, pulmonary hypoplasia, gall-bladder agenesis, congenital diaphragmatic hernia and club hand and clubfoot have also been reported prenatally. Their frequency varies from 10% to 20% [11].
Several studies have underlined the importance of a strong genotype-phenotype correlation in WHS [8,12]. Thus, three different phenotypes are usually described: Micro deletions less than 3.5 Mb in size are associated with a mild phenotype represented by a typical facial appearance without microcephaly and growth retardation. Deletions between 5 and 18 Mb are associated with the classical WHS phenotype, represented by a severe psychomotor retardation. For deletions larger than 22 Mb, a severe phenotype with major malformations (most likely incompatible with life) is observed [8,11]. Battaglia et al. [13] analyzed more than 100 cases and demonstrated that a submicroscopic deletion, which was detected only by molecular techniques, could result in a severe WHS phenotype. They concluded that there is no strict correlation between deletion size and phenotype, mostly due to balanced rearrangements.
A minority of rearrangements arises from a parental balanced translocation, not more than 10-14%. The majority, about 85-90%, are de novo. However, de novo rearrangements, all expected to be isolated deletions, are actually isolated deletions in 70% of cases, the remaining are complex rearrangements, above all unbalanced de novo translocations. Thus, 40-50% of the rearrangements are unbalanced translocations, however most of them are de novo. This aspect is very important for the proper genetic counseling and genetic testing to be offered to the families.
Most cases of prenatal diagnosis of WHS published in the medical literature have been performed through conventional cytogenetic analysis. During the last decade, the availability of new techniques, particularly Comparative Genomic Hybridization Array (CGH-array), has allowed a much more accurate description of the molecular mechanisms leading to WHS, including the diagnosis of complex phenotypes associated with submicroscopic deletions [12,14]. A study of 21 patients with SWH phenotype by Maas et al. [10] revealed that 8 patients had a deletion detectable by conventional cytogenetic techniques while 13 had a submicroscopic deletion that could only be diagnosed by microarray.
In conclusion, growth restriction as an isolated finding or associated with facial dysmorphism and other major malformations may be suggestive of WHS, and should trigger genetic investigation. A combined diagnostic approach based on conventional karyotyping and molecular analysis can offer a quick and definitive result to allow accurate prognoses. Parents must be studied by conventional cytogenetics and by FISH to search for a parental balanced translocation that allow genetic counseling for the family.
Acknowledgments
We would like to thank to Departament of Obstetrics and Gynecology Virgen de las Nieves University Hospital for providing the figures and the family for their assistance with this case.
References
-
Titomanlio L, Romano A, Conti A, Genesio R, Salerno M, et al. (2004) Mild Wolf-Hirschhorn phenotype and partial GH deficiency in a patient with a 4p terminal deletion. Am J Med Genet A 127A: 197-200.
-
Wolf U, Reinwein H, Porsch R, Schr�ter R, Baitsch H (1965) [Deficiency on the short arms of a chromosome No. 4]. Humangenetik 1: 397-413.
-
Hirschhorn K, Cooper HL, Firschein IL (1965) Deletion of short arms of chromosome 4-5 in a child with defects of midline fusion. Humangenetik 1: 479-482.
-
Battaglia A, Filippi T, Carey JC (2008) Update on the clinical features and natural history of Wolf-Hirschhorn (4p-) syndrome: experience with 87 patients and recommendations for routine health supervision. Am J Med Genet C Semin Med Genet 148C: 246-251.
-
Sifakis S, Manolakos E, Vetro A, Kappou D, Peitsidis P, et al. (2012) Prenatal diagnosis of Wolf-Hirschhorn syndrome confirmed by comparative genomic hybridization array: report of two cases and review of the literature. Mol Cytogenet 5: 12.
-
Bergemann AD, Cole F, Hirschhorn K (2005) The etiology of Wolf-Hirschhorn syndrome. Trends Genet 21: 188-195.
-
Engbers H, van der Smagt JJ, van 't Slot R, Vermeesch JR, Hochstenbach R, et al. (2009) Wolf-Hirschhorn syndrome facial dysmorphic features in a patient with a terminal 4p16.3 deletion telomeric to the WHSCR and WHSCR 2 regions. Eur J Hum Genet 17: 129-132.
-
Zollino M, Murdolo M, Marangi G, Pecile V, Galasso, et al. (2008) On the nosology and pathogenesis of Wolf-Hirschhorn syndrome: genotype-phenotype correlation analysis of 80 patients and literatura review. Am J Mec Genet C Semin Med 148C: 257-269.
-
Chen CP, Su YN, Chen YY, Sun JW, Chern SR, et al. (2011) Wolf-Hirschhorn (4p-) syndrome: Prenatal diagnosis, molecular cytogenetic characterization and association with a 1.2-Mb microduplication at 8p22p21.3 and 1.1- Mb microduplication at 10p15.3 in a fetus with an apparently pure 4p deletion. Taiwanese J Obstet Gynecol 50: 506-511.
-
Maas NM, Van Buggnhout G, Hannes F, Thienpont B, Sanlaville D, et al. (2008) Genotype-phenotype correlation in 21 patients with Wolf-Hirschhorn syndrome using high resolution array comparative genome hybridization (CGH). J Med Genet 45: 71-80.
-
Debost-Legrand A, Goumy C, Laurichesse-Delmas H, D�chelotte P, Beaufr�re AM, et al. (2013) Prenatal ultrasound findings observed in the Wolf- Hirschhorn syndrome: data from the registry of congenital malformations in Auvergne. Birth Defects Res A Clin Mol Teratol 97: 806-811.
-
Chao A, Lee YS, Chao AS, Wang TH, Chang SD (2006) Microarray-based comparative genomic hybridization analysis of Wolf-Hirschhorn syndrome in a fetus with deletion of 4p15.3 to 4pter. Birth Defects Res A Clin Mol Teratol 76: 739-743.
-
Battaglia A, Carey JC, Wright TJ (2001) Wolf-Hirschhorn (4p-) syndrome. Adv Pediatr 48: 75-113.
-
Rosell� M, Monfort S, Orellana C, Ferrer-Bolufer I, Quiroga R, et al. (2009) Submicroscopic duplication of the Wolf-Hirschhorn critical region with a 4p terminal deletion. Cytogenet Genome Res 125: 103-108.
