

Obstetrics and Gynaecology Cases - Reviews
Case Report: 7q11 Microdeletion in Swyer Syndrome Patient
Patrick T Reeves1, Axel G Moreira1, Tochi M Amagwula2, Toy G Lee2, Dominique D Washington2, Akila Muthukumar3 and John Y Phelps2*
1School of Medicine, The University of Texas Medical Branch at Galveston, USA
2Department of Obstetrics & Gynecology, The University of Texas Medical Branch at Galveston, USA
3Department of Pediatrics, The University of Texas Medical Branch at Galveston, USA
*Corresponding author: John Y. Phelps, Associate Professor, Department of Obstetrics & Gynecology, The University of Texas Medical Branch at Galveston, 3,120 John Sealy Annex, Galveston, TX- 77555-0587, USA, Tel: 409-772-8598, Fax: 409-747-5129, E-mail: jyphelps@utmb.edu
Obstet Gynecol Cases Rev, OGCR-2-036, (Volume 2, Issue 3), Case Report; ISSN: 2377-9004
Received: March 28, 2015 | Accepted: May 01, 2015 | Published: May 04, 2015
Citation: Reeves PT, Moreira AG, Amagwula TM, Lee TG, Washington DD, et al. (2015) Case Report: 7q11 Microdeletion in Swyer Syndrome Patient. Obstet Gynecol Cases Rev 2:036. 10.23937/2377-9004/1410036
Copyright: © 2015 Reeves PT, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Introduction: Swyer syndrome or XY gonadal dysgenesis is caused by genetic mutations in testis-determining factors, such as the SRY gene, that lead to streak gonads in phenotypical females. The following case demonstrates a patient with a 7q11 microdeletion, a region which governs the coding of a transcription regulator called Zinc Finger 92 (ZNF92).
Case: A 13-year-old female presented to the clinic with an abdominal mass, 30lb weight loss, and increased urinary frequency. After further work up, the abdominal mass was identified as a dysgerminoma. Genetic testing of the patient revealed an XY genotype, with an intact SRY region, as well as a deletion at 7q11.22.The 7q11 region contains both Zinc Finger 92, a region noted for prominent RNA transcription regulation, and a prominent tumor suppressor gene.
Conclusions: Previously established mechanisms have suggested improper encapsulation of germ cells within dysgenic gonads and increased Bcl-2 expression can lead to dysgerminoma formation. Additionally, studies suggest the presence of a tumor suppressor gene is located on 7q since deletions in that region have been linked to numerous ovarian neoplasms. Thus, it is possible the gonadal dysgenesis experienced by this patient was caused by a 7q11 deletion and loss of ZNF92. By extension, this mechanism for gonadal dysgenesis may have exacerbated aberrant germ cell growth and dysgerminoma development in the setting of tumor suppressor gene loss from 7q. However, further research is required to determine if a microdeletion in 7q11 is associated with Swyer syndrome.
Keywords
Swyer, Dysgerminoma, Gonadal dysgenesis, Zinc finger, SRY, 7q11
Introduction
Swyer syndrome is an embryologic disorder manifested by intact M�llerian organs and primary amenorrhea with the absence of secondary sexual characteristics. With an incidence of 1:80,000 births, the hallmark of Swyer syndrome is the presence of dysfunctional ovarian tissues [1]. Among patients with Swyer syndrome, 5% will develop dysgerminoma-the most common type of ovarian germ cell tumor [2]. Gonadal dysgenesis is responsible for an increased risk of dysgerminoma due to failed encapsulation of germ cells within primordial follicles at birth. Germ cells can escape this encapsulation process and avoid termination through intraovarian expression of Bcl-2 [3,4]. As a result, they can undergo rapid, unchecked growth resulting in malignant tumor development [5-7]. Due to this risk, the American College of Obstetrics and Gynecology recommends prophylactic bilateral gonadectomy for all patients diagnosed with gonadal dysgenesis. Recent studies have demonstrated a higher incidence of ovarian cancer among women who possess a regional deletion on chromosome 7q, which has provided researchers with the hypothesis that a tumor suppressor gene (TSG) is normally located on the short arm of chromosome 7 and can be lost by sporadic deletion [8,9]. Thus, while the risk of developing dysgerminoma in conjunction with Swyer syndrome linked to aberrant germ cell growth is clearly established, a compounded risk may be held with Swyer patients possessing 7q deletions due to loss of a TSG.
Here we present a unique case concerning a specific genetic anomaly that may both predispose a patient to developing Swyer syndrome and function as a sensitive risk factor for the development of dysgerminoma. While it is generally known that deletion of the SRY region on the Y chromosome can cause Swyer syndrome, there are other various sporadic mutations that may affect the embryologic development of testes and cause this syndromic presentation [10-13]. The patient in this case presentation did not have a microdeletion in the SRY gene as is typically seen in patients with Swyer syndrome. In this case, a female patient who appeared phenotypically normal possessed an XY karyotype, a normal SRY region, and a 443 kilo base pair deletion at 7q11.21. This region, which governs the coding of a transcription factor regulator called Zinc Finger 92 (ZNF92), shows moderate to strong RNA expression in the gonads without affecting uterine growth, possibly leading to sexual developmental disorders [10-14]. This hypothesis parallels the presentation of our patient who possesses a presumed uterus as well as dysfunctional gonads coinciding with a deletion of this transcription regulator region on chromosome 7q [10-12]. Mechanistically, this case of Swyer syndrome stemmed from dysregulated RNA transcription of gonadal tissue. Through the loss of ZNF92, we have theorized that this patient lacked the proper gene regulators to ensure robust, adequate transcription and gonadal development. Thus, by losing 7q11.22, the patient developed dysfunctional gonads which could have led to poor encapsulation of germ cells within primordial follicles. The additional loss of a prominent tumor suppressor gene may have placed this patient at an above average risk of malignant germ cell transformation with concern to the diagnosis of Swyer syndrome.
Case Study
Patient is EG, a 13-year-old female who presented to the Pediatrics clinic with diffuse abdominal pain. She reported a 30-lb weight loss in the past 6 months along with decreased appetite, nausea, and increased urinary frequency. On physical exam, a hard, palpable lower abdominal mass was noted. Developmentally, Tanner Stage 1 findings consistent with absence of glandular collections around the areola, a lack of areolar elevation from skin contours, as well as a paucity of pubic and axillary hair were noted. There were no physical manifestations of excess androgenicity or virilism present (ie, acne, hirsuitism, striae, or alopecia). The patient was admitted to pediatric services for evaluation using multiple radiographic modalities, including Computed Tomography (CT) scan and abdominal ultrasound. The CT revealed a 20x16x9cm multilobulated solid mass with focal areas of calcification along with moderate hydronephrosis bilaterally (Figure 1). The radiology report stated that �tumor implants vs enlarged lymph nodes� were seen along iliac vessels with the largest measuring approximately 2 cm. On admission, serologic testing revealed an elevated creatinine of 1.39mg/dL, likely caused by bilateral hydronephrosis from mass effect. A bone scan was performed which demonstrated no evidence of malignancy or osteopenia. An abdominal ultrasound demonstrated a uterus measuring 4.19cmx2.27cm. This size is consistent with a prepubertal, small uterus commonly found in Swyer syndrome [15]. Further laboratory diagnostics used to explore various causes of primary amenorrhea and thus narrow the differential are depicted in Table 1.
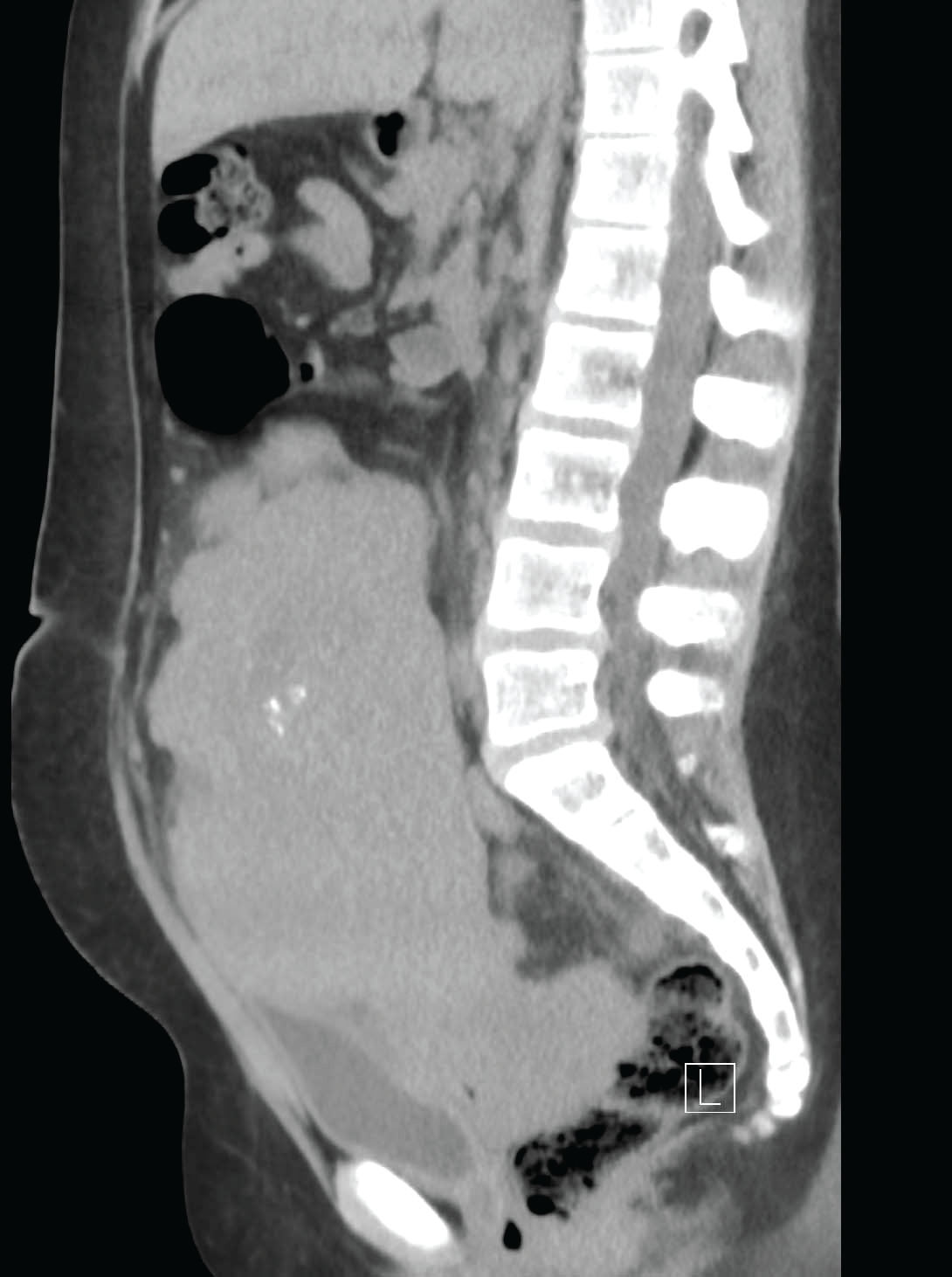
.
Figure 1: Sagittal view of CT abdomen/pelvis demonstrating large solid mass in pelvis of Swyer patient.
View Figure 1
![]()
Table 1: Laboratory results for Swyer patient.
View Table 1
During the primary admission, the pediatric surgeon retrieved an intraoperative frozen biopsy from the abdominal mass that confirmed histological findings consistent with dysgerminoma. This result was well correlated with the elevated serum lactate dehydrogenase (LDH) found in the patients serum, which is characteristic of a dysgerminoma [5]. The pediatric surgeon also performed a vaginoscopy at the time of the operation that revealed normal, external female genitalia and anatomical architecture. Further evaluation of the biopsy specimen revealed neoplastic cells positive for CD117 (ckit) and Oct3/4, classic for a dysgerminoma [5]. These cells were negative for WT-1, AE1/3, and CD45. Biopsy sample cytogenetics revealed an XY genotype. This result led the team to perform a chromosomal microarray analysis of the patient's peripheral blood. This test definitively confirmed the XY makeup, with a 443 kbp loss on 7q11.Gynecologic oncology was subsequently consulted and has recommended the patient have a mass removal with bilateral gonadectomy.
Discussion
Patients with Swyer syndrome tend to present to their primary care provider in their teens with primary amenorrhea, unambiguous external genitalia, and a lack of axillary and pubic hair [1]. The presence of normal female levels of testosterone and DHT and a lack of secondary sexual characteristics is pathognomonic for Swyer Syndrome. This disorder must be differentiated from congenital androgen insensitivity syndrome, which is exhibited by enhanced breast development with minimal axial army and pubic hair in the setting of increased levels of testosterone and DHT. In addition, 5-alpha reductase deficiency (5-ARD) should be considered in the differential of primary ammenorhea, especially when physical findings are suggestive of androgen insensitivity. This patient was seen for a well-child visit shortly before diagnosis at which time no pertinent findings were noted except delayed puberty. Diagnosis of the patient with Swyer syndrome can be challenging. The differential diagnosis in this patient included Turner syndrome, androgen insensitivity syndrome (AIS), 5-ARD, and Mullerian agenesis (Mayer Rokitansky Kuster Hauser syndrome). This patient did not have the characteristic short stature of Turners and lacked secondary sexual characteristics found in AIS. Both the presence of a rudimentary uterus as well as the patient's subnormal levels of DHT and testosterone helped to rule out 5-ARD and AIS. Finally, the finding of low Anti-Mullerian Hormone was not consistent with Mullerian agenesis. Peripheral blood karyotype, chromosomal microarray, and pathology specimens were consistent with XY karyotype, and imaging demonstrated presumed internal female organs. Karyotype and clinical presentation therefore led to the diagnosis of Swyer syndrome. This patient did not demonstrate a microdeletion in the SRY gene but a deletion at 7q11. Moreover, the SRY region appeared completely intact without point mutations or other genetic abnormalities. Further evaluation of the chromosomal microarray did not reveal any evidence to alteration of WT-1 or other sex-determining regions. Similar to this case, molecular epidemiologic studies conducted by Sohal have identified novel microdeletions of 7q11, which can be linked to disordered RNA transcription within various stromal lineages [13]. In addition, Peoples et al previously identified novel transcription regulators on 7q which, when lost, can contribute to complex developmental phenotypes including Williams syndrome.
Gonadal dysgenesis is associated with dysgerminoma development. Failed encapsulation of germ cells with subsequent overexpression of Bcl-2 can lead to aberrant cell growth and malignant transformation. However, with respect to this unique presentation of Swyer�s, it is important to note that deletions of the region 7p12�q11 have been linked to the development of various ovarian neoplasms, including serous epithelial neoplasms, ovarian adenocarcinomas and choriocarcinomas [8,9,16]. Studies have revealed that a loss in this region is correlated with such a high risk of cancer development that a tumor suppressor gene is likely located on 7q [8,16]. The increased expression of ZNF92 in T lymphoid cells further supports the study conducted by Zhao et al. demonstrating T lymphocyte accumulation in dysgerminomas and seminomas [17,18]. Thus, the patient�s deletion at 7q11 may have caused not only her Swyer Syndrome presentation but also may have precipitated the malignant transformation of germ cells by eliminating a tumor suppressor that would have provided protection against ovarian neoplasm development. This genetic phenomenon requires more research to ascertain whether true causality exists between the transcription regulator and the presentation of Swyer syndrome.
References
-
Michala L, Goswami D, Creighton SM, Conway GS (2008) Swyer syndrome: presentation and outcomes. BJOG 115: 737-741.
-
Behtash N, Karimi Zarchi M (2007) Dysgerminoma in three patients with Swyer syndrome. World J Surg Oncol 5: 71.
-
Kovacs, William J, Ojeda SR (2011) Textbook of endocrine physiology. Oxford University Press, 2011.
-
Begum H, Roy CR, Moniruddin ABM (2008) Case history: A healthy pregnancy following chemotherapy for dysgerminoma. The ORION: 539-540.
-
Kumar V, Abbas AK, Aster JC (2014) Robbins and Cotran. Pathologic basis of diseass. (Professional Edn). Expert Consult-Online. Elsevier Health Sciences.
-
Sanfilippo, Joseph S (2001) Pediatric and adolescent gynecology. WB Saunders Company.
-
Talerman A, Vang R (1994) Germ cell tumors of the ovary. Blaustein�s pathology of the female genital tract. Springer, New York: 849-914.
-
Neville PJ,Thomas N, Campbell IG (2001) Loss of heterozygosity at 7q22 and mutation analysis of the CDP gene in human epithelial ovarian tumors. Int J Cancer 91: 345-349.
-
Kerr J, Leary JA, Hurst T, Shih YC, Antalis TM, et al. (1996) Allelic loss on chromosome 7q in ovarian adenocarcinomas: two critical regions and a rearrangement of the PLANH1 locus. Oncogene 13: 1815-1818.
-
GNC, Entrez Gene, and UniProtKB/Swiss-Prot. ZNF92 Gene. � Gene Cards. Life Map, n.d. Web.
-
NCBI, NIH (2010) Homo Sapiens Complex Locus ZNF9, Encoding Zinc Finger Protein 92. NCBI. U.S. National Library of Medicine.
-
Peoples RJ, Cisco MJ, Kaplan P, Francke U (1998) Identification of the WBSCR9 gene, encoding a novel transcriptional regulator, in the Williams-Beuren syndrome deletion at 7q11.23. Cytogenet Cell Genet 82: 238-246.
-
Sohal D, Opalinska J, Zhou L, Pahanish P, Friedman E, et al. (2007) High Resolution Epigenomic Profiling of Loss of Heterozygosity in MDS Reveals an Important Role of DNA Methylation in Regulating Expression of Genes in the Deleted Regions of Chromosomes 5q, 7q and 20q. ASH Annual Meeting Abstracts 110: 11.
-
Polityko AD, Khurs OM, Kulpanovich AI, Mosse KA, Solntsava AV, et al. (2009) Paternally derived der(7)t(Y;7)(p11.1 approximately 11.2;p22.3)dn in a mosaic case with Turner syndrome. Eur J Med Genet 52: 207-210.
-
Han Y, Wang Y, Li Q, Dai S, He A, et al. (2011) Dysgerminoma in a case of 46, XY pure gonadal dysgenesis (Swyer syndrome): a case report. Diagn Pathol 6: 84.
-
Burke B, Sebire NJ, Moss J, Hodges MD, Seckl MJ, et al. (2006) Evaluation of deletions in 7q11.2 and 8p12-p21 as prognostic indicators of tumour development following molar pregnancy. Gynecol Oncol 103: 642-648.
-
Bellefroid EJ, Marine JC, Ried T, Lecocq PJ, Rivi�re M, et al. (1993) Clustered organization of homologous KRAB zinc-finger genes with enhanced expression in human T lymphoid cells. EMBO J 12: 1363-1374.
-
Zhao X, Wei YQ, Kariya Y, Teshigawara K, Uchida A (1995) Accumulation of gamma/delta T cells in human dysgerminoma and seminoma: roles in autologous tumor killing and granuloma formation. Immunol Invest. 24: 607-618.
