

International Journal of Allergy Medications
HAE in Children- What is the Best Treatment Strategy?
Carlos Alberto Sanchez Salguero* and Alvaro Isidro Sanchez Chacon
Pediatric Department, School of Medicine, University Hospital Puerto Real, Cadiz, Spain
*Corresponding author:
Carlos Sanchez Salguero, Head of Pediatric Allergy and Pneumology Section, Professor of Pediatric, School of Medicine, University Hospital Puerto Real, Cadiz, Spain, E-mail: libraygeminis@hotmail.com
Int J Aller Medcations, IJAM-2-016, (Volume 2, Issue 1), Mini Review
Received: March 05, 2016: Accepted: April 28, 2016: Published: April 30, 2016
Citation: Salguero CAS, Chacon AIS (2016) HAE in Children- What is the Best Treatment Strategy? Approval of a Drug. Int J Aller Medcations 2:016.
Copyright: © 2016 Salguero CAS, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
The hereditary angioedema due to C1 inhibitor deficiency (C1 esterase inhibitor), is classified in two types and is considered a disease, included in the group of rare diseases, which usually occurs during childhood and adolescence with intermittent attacks of angioedema that can endanger the patient's life. The diagnosis should be possible as early as we should avoid establishing ineffective treatments, and always seek the best treatment for inflammation. There have been several expert meetings on the subject, the most important being sponsored by the WAO in 2012 and subsequently publishing the International Working Group, although the responsible medical for treating a patient with HAE can use other usage guidelines. Of all the treatments available in Pediatrics one can conclude that the concentrate of C1-inhibitor derived from plasma is considered the best treatment option for this patient group. The case presented in the discussion section is a patient in whom it begins to use Berinert but finally, due to the intensity of the attack in addition to possible drug resistance, opted for a combination treatment Berinert-Cinryze that was the most adequate, and there is currently no publication on their combined use.
Keywords
C1-INH (C1 inhibitor, C1-esterase inhibitor), Hereditary angioedema, Pediatric, Plasma-derived C1-INH concentrate
Abbreviations
C1-INH: C1 inhibitor; AE: angioedema; HAE: Hereditary angioedema; HAE-C1-INH: Hereditary angioedema due to C1 esterase inhibitor deficiency; pDC1-INH: plasma-derived C1-INH concentrate
Introduction
Hereditary angioedema is a rare disease that occurs in children and adolescents. It was first described by Quincke in 1882 and in 1888 Osler made a full clinical description [1]. The pathogenesis is based on insufficient levels of C1 inhibitor in HAE type 1; or due to the presence of a C1 -inhibitor with altered function despite having normal serum, or type 2 HAE. There is also a more typical female type 3 HAE, characterized by normal levels of C1-INH functional although with a mutation in F12 gene, this type III is rare in children and adolescents.
HAE is a genetic disease with autosomal dominant transmission and an estimated prevalence 1: 50000 [2] without differences in gender or race predominance. It is defined as recurrent episodes of angioedema no pruritic that can affect any part of the body, typically located in upper and lower extremities, gastrointestinal tract, face and airways [3]. The disease begins in childhood, about 2-3 years and over 50% develop symptoms at 10 years, worse in adolescence and persist throughout life.
The HAE is the result of mutations in the C1 inhibitor gene located on chromosome 11 (p11.2-q13) [4]. Genetic studies have identified more than 300 mutations found a clear correlation between the genetic defect and severity of the disease. The type I would relate to abnormal proteins or alterations in the processing of proteins; whereas type II is related to a defective protein with a mutation in exon 8. And this despite 25% of patients do not have family history because are spontaneous mutations.
Pathophysiology
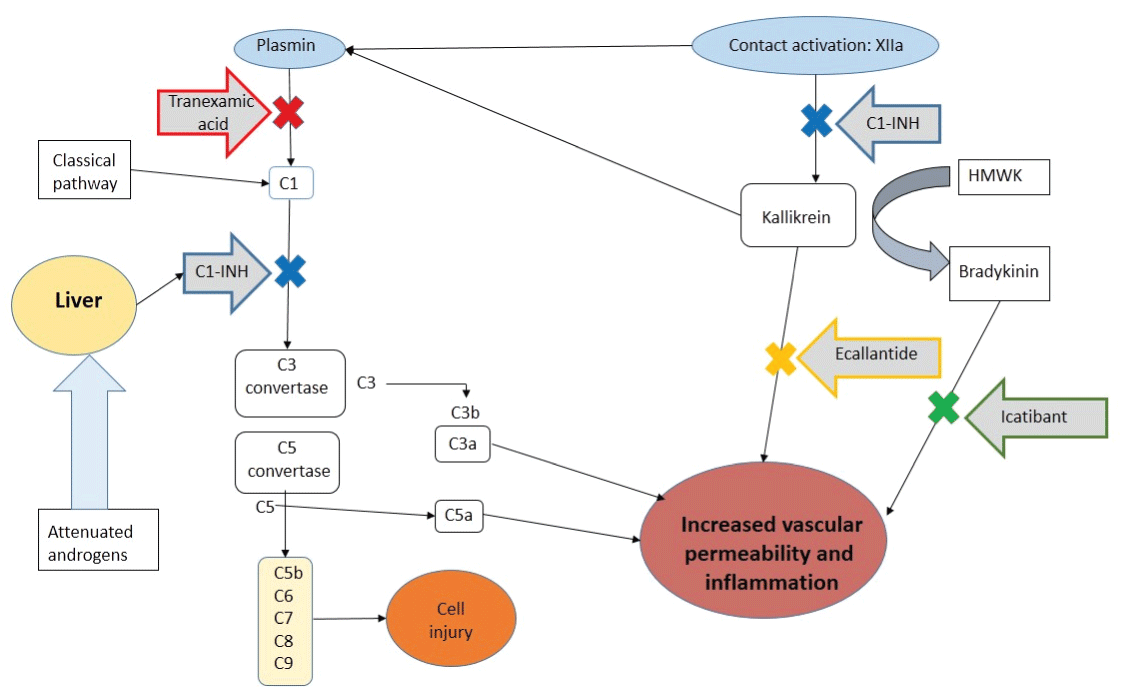
C1 inhibitor is a member of the family of serpins or serine protease inhibitors, SERPING 1 is the name of the gene [5]. It is a protein synthesized in the liver and represents the main protease inhibitor of complement (C1r, C1s, MASP-1 and MASP-2) and proteases of the contact system. It is also less protease inhibitor of the fibrinolytic system and coagulation [4,6].
Although the majority of patients with HAE are heterozygous, there are also homozygous cases reported. Normally is only affected one allele, which should then expect a level of inhibitor of at least 50% of its normal value, however these patients show levels 30-50%. This is attributed to the mechanism of action of serpins, which form a serpin protease in which both are inactivated and are degraded complex. When consumed C1-INH in the various ways in which it participates, presenting consumed lower levels than expected [5].
In patients with type II disease, the levels are normal or elevated as the default function of the protein half-life is prolonged unable to form enzyme complexes [5].
Figure 1 shows the coagulation cascade, the complement and contact systems and sites in which C1-INHis involved. Targets are XIIa and XIa factors, C1r, C1s, MASP-1 and MASP-2 factors of the classical pathway and the lectin respectively and kallikrein, important mediator responsible for the generation of bradykinin from high molecular weight kininogen (HMWK) [2,6]. The main mediator of angioedema is bradykinin; due to the absence of the inhibitor, the consumed complement cascade is activated and consumed C2 and C4. Fibrinolytic pathway also involved in preventing the conversion of plasminogen to plasmin [2,5,6].
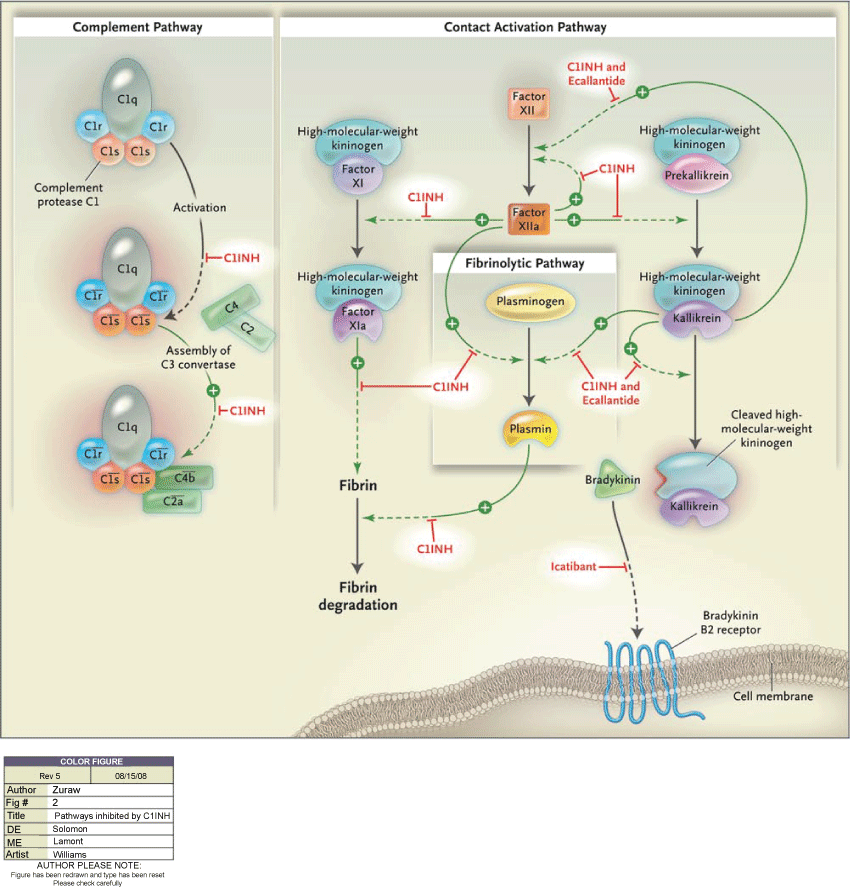
In the classic complement pathway, the complement protease C1 is activated and then assembles the C3 convertase. (Activation is indicated by horizontal bars over the complement names). In the contact activation pathway, trace amounts of factor XIIa activate additional factor XII, as well as prekallikrein. Activated factor XIIa activates factor XI to factor XIa, leading to enhanced fibrin formation. Activated factor XIIa and kallikrein activate each other, and then plasma kallikrein cleaves high-molecular-weight kininogen to release bradykinin.
In the fibrinolytic pathway, plasminogen is activated to plasmin, which cleaves fibrin. Proteolytic activities are indicated with green arrows and point toward the steps they catalyze. Steps inhibited by C1INH, through conventional or new types of therapy, or by two other new drugs being investigated for the treatment of hereditary angioedema are shown with red T bars [7].
Kinins are peptides that stimulate relaxation of vascular smooth muscle and increases vascular permeability, being interesting the role of bradykinin, primary mediator responsible angioedema, which exerts its biological action through the B2 receptor expressed in cells and vascular smooth muscle cells of endothelium. In humans there are other B1 receptor expressed in tissues after injury or upon contact with bacterial endotoxins, which is also involved in the onset of angioedema associated with physical trauma or infection. Bradykinin degradation is carried out by kininases I and II known as carboxypeptidases N and ACE, and other enzymes such as DPP IV.
Clinical Picture
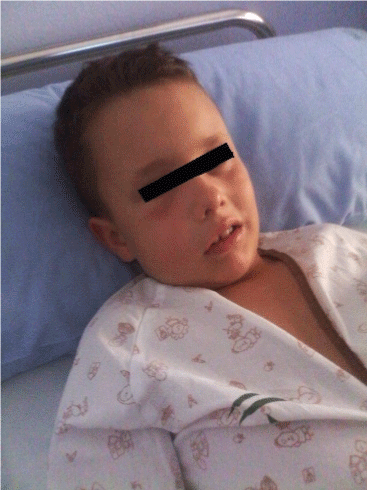
Prodromal symptoms may occur up to 1/3 of cases [8]. and include numbness and marginalized erythema and pruritic serpiginous no rush. Edema typically develops over 24 hours and yields in the next 48-72 hours, affecting mainly the face, arms, legs, hands, feet and abdomen [1]. It can occur in a body region, then migrate to another before resolving or even affect multiple sites simultaneously. It is important to highlight the absence of urticarial which is a characteristic of this disease (Figure 2).
More than 90% of patients present with an abdominal episode sometimes [9]. The pain is intense, accompanied by diarrhea, nausea and vomiting. The bowel sounds are diminished or absent and sometimes simulate an acute abdomen. During these crises there is a passage of interstitial fluid into the peritoneal cavity which can cause hypotension and some patients undergo exploratory surgery.
Laryngeal edema poses a risk to the patient's life, occurring in 50% of patients with a mortality rate of 30% [2,10] (Figure 3).
Anything can trigger the crisis, but the most common are: physical trauma, surgical, medical and dental, prolonged standing, infections, emotional stress and certain drugs [2,6,11].
Diagnosis
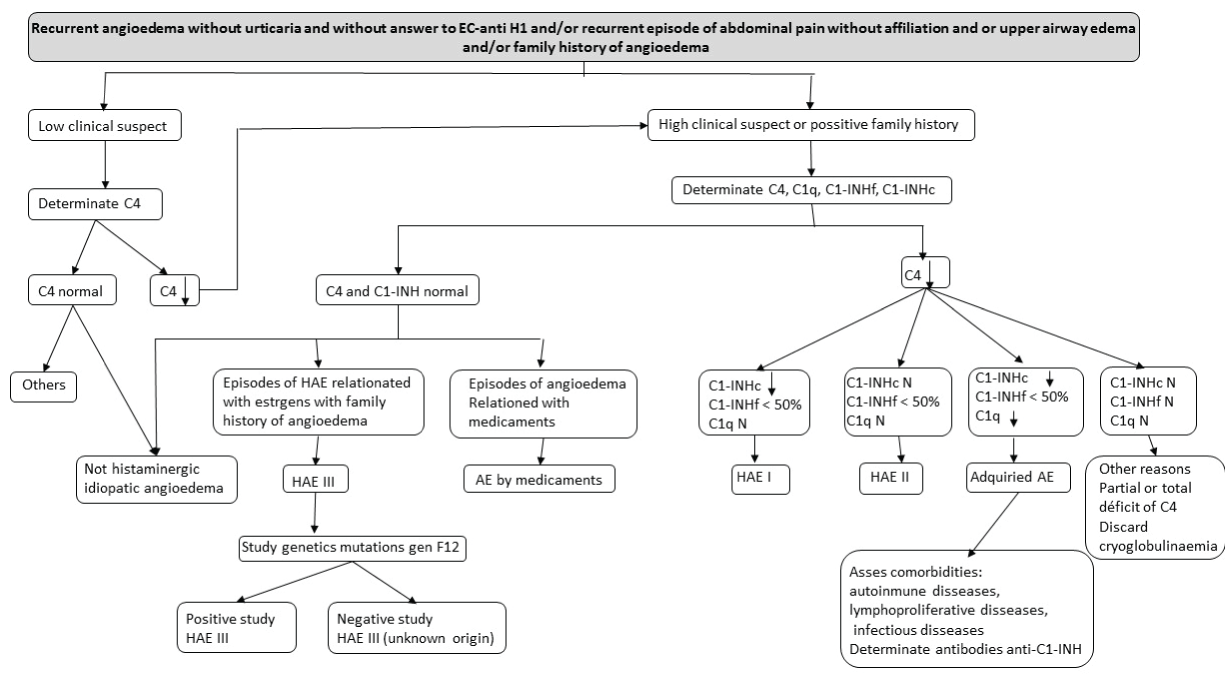
It is based on recognizing the symptoms, family history (though there is a 20% de novo) and laboratory data. The first sign to consider are repeated episodes of angioedema (edema repeated joints lead to confusion with systemic autoimmune diseases) [12,13].
We have taken a decision algorithm for the differential diagnosis of HAE mediated by bradykinin, after the appearance of recurrent angioedema, recurrent episodes of abdominal pain without affiliation upper airway edema and/or family history of angioedema. This algorithm had been published in JACI 2011 by Caballero et al. [12].
Recurrent angioedema is defined as the presence of more than one episode of acute self-limiting angioedema with symptom-free periods, regardless of their severity and location [1,2,12]. Also unanswered angioedema corticosteroids H1-antihistamines, acute episodes that do not respond to doses of methylprednisolone ≥ 1 mg/kg or other equipotent dose corticosteroids or recurrent AE fails to prevent that daily treatment with anti-H1 dose high or corticosteroids [14].
Treatment
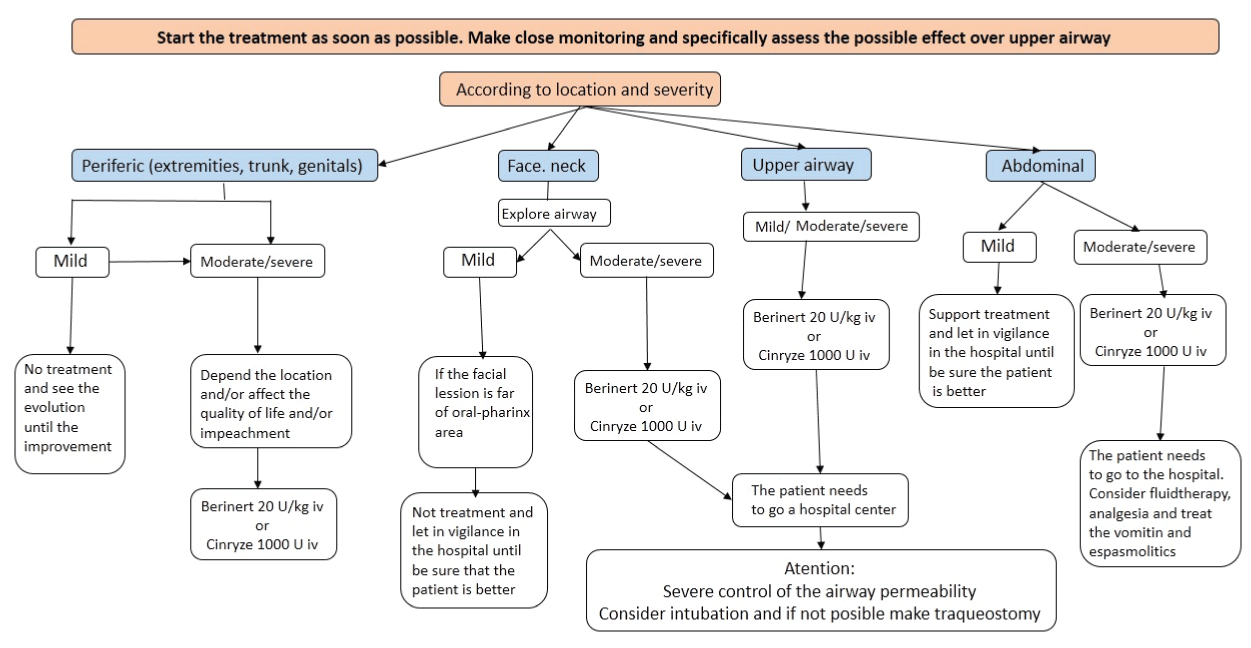
When a patient has unmistakable symptoms of an acute attack of AE, treatment should be started early if the clinical situation warrants, conduct intensive monitoring of the patient and assess the possible occurrence of involvement of the upper airway. The decision whether to treat an acute attack are set depending on the location and severity of edema.
The peripheral edema (limbs, trunk and genitals), if they are mild and self-limiting may not require treatment, like the face or neck if they are mild and are far from the oropharynx. If impairment of quality of life, severity and/or disability for daily activity of the patient, should be considered the treatment of acute episode. If acute attacks recur frequently it should consider the long-term prophylaxis. On the other hand, the upper airway edema can cause suffocation and death, so it should be treated even in mild cases. It is usually necessary to go to hospital for a strict control of airway permeability, as sometimes intubation, which must be done by a professional. Sometimes it is necessary to perform an emergency tracheotomy.
In the abdominal acute attacks mild should relieve the pain first and kept under observation to objectify clinical improvement, but if it is moderate or severe, you should go to the hospital where, in addition to the treatment of angioedema, should consider using fluid, antiemetics, analgesics and antispasmodics.
In 2010 some recommendations were issued by consensus for therapeutic strategy in all age groups [15], and had previously published a consensus in the pediatric population [16].
![]()
Table 1: Approval status of products for the treatment of HAE-C1-INH (Europe and USA) < 18 years old.
View Table 1
Table 1 shows an overview of all drugs approved for treatment of AE. Specific risks and adverse effects of the treatments available are set out in table 2 and table 3 have drugs for pediatric use.
Human plasma-derived C1-INH (Pd C1-INH) concentrate. Human C1-INH is a glycoprotein containing 478 amino acids with an approximate molecular weight of 105 kDa. It is a family member of the serine protease inhibitor (serpin) and its main function is to inhibit drug early activation steps of the complement system and the kallikrein-kinin.
![]()
Table 2: Specific risks and adverse effects of products for the treatment of HAE-C1-INH.
View Table 2
![]()
Table 3: Assessment on pediatric use for the treatment of HAE-C1-INH.
View Table 3
Berinert®
It is derived from human plasma. The recommended dose is 20 U/kg, intravenously, and is approved for use in all pediatric age groups. There have been several studies on the main dose, but the most decisive have been the IMPACT-1 and its extension the IMPACT-2. In the IMPACT-1 included 124 patients (aged 6-72 years) treated in every crisis of angioedema (abdominal or facial) with 10 U/kg, 20 U/kg or placebo. It was found as to the dose of 20 U/kg the mean time to response was much less than with placebo (0.5 vs. 1.5 hours, p = 0.0025), whereas this time was not statistically significant with dose of 10 U/kg (1.2 hours). Also with 20 U/kg medium for complete resolution of HAE time it was significantly shorter compared to placebo (4.92 vs. 7.79 hours, p = 0.0237) [17].
Subsequently the study was extended in the IMPACT-2 investigating the long-term therapy with Berinert, including 1805 attacks in any area of the body in 57 patients (aged 10-53 years) and treated with 20 U/kg of Berinert [18]. The mean time to onset of response to symptoms was 0.46 hours and the average time to complete resolution of all symptoms was 15.5 hours (shorter in cases of laryngeal involvement was 5.8 hours). In 99% of the attacks it was only necessary to use 20 U/kg, without occurrence of adverse effects without anti-C1-INH antibodies detected. Finally, there are more recent studies that demonstrate the effectiveness and safety of Berinert in children and adolescents [19,20].
Cinryze®
It is derived from human plasma, obtained by nanofiltration. The recommended dose is 1000 U (2 vials), intravenously regardless of patient weight. Cinryze was approved for use in adolescents with acute attacks of HAE and for long-term prophylaxis.
They have published several studies use but we highlight two double-blind, randomized, placebo-controlled; one for use in another acute attacks and other in long term prophylaxis, without noticeable adverse effects.
The first study included 71 patients with acute attacks of inflammation affecting face, abdomen or external genitalia treated with 1000 U of Cinryze or placebo. Patients who did not respond in one hour received a second dose. If the symptoms subsided after 4 hours, the patient received another dose of 1000 U. The average response time was 2 hours Cinryze (placebo 4 hours; p = 0.048); with a mean time to complete resolution of symptoms Cinryze 12.3 hours (25 hours placebo; p = 0.004) [21].
In the study of long-term prophylaxis were included 24 patients obtained of the previous study and with at least two attacks per month. They received 1000 U Cinryze or placebo every 3-4 days (crossover after 12 weeks). During the 12 weeks of treatment, the number of inflammatory processes was significantly lower with Cinryze compared to placebo (6.1 vs. 12.7 attacks; p < 0.0001) [22].
Numerous studies of patients under 18 years treated with Cinryze (2-5 years: 2 patients; 6-11: 17 patients; 12-17: 26 patients) [23].
Ruconest®
It is a recombinant C1-INH concentrate human. The active molecule, conestat alpha is a recombinant analogue of the human C1-INH (glycoprotein with a molecular weight of 71 kDa) whose glycosylation is not identical to human C1-INH, which is why its half-life is shorter [24]. Ruconest is derived from the recombination of milk produced and purified from transgenic rabbits of New Zealand. The recommended dose is 50 U/kg intravenously (with a maximum dose of 4200U in patients weighing > 84 kg, up to two doses in 24 hours).
Include two randomized, double-blind, placebo-controlled (100 U/kg vs. placebo and 50 U/kg vs. 100 U/kg vs. placebo) [25]. The remission of symptoms was similar and significantly faster with a dose of 50 U/kg and 100 U/kg compared to placebo. No adverse effects were observed with use although there are no data of indication in pediatric indication. Kallikrein inhibitors and receptor antagonists of bradykinin.
Icatibant-Firazyr®
It is a decapeptide chemically synthesized with a selective B2 bradykinin receptor antagonist, resistant to enzymatic degradation. The recommended dose is 30 mg, subcutaneously, with a maximum of 3 doses in 24 hours. There is no safety profile in children so its use in pediatric patients is not permitted.
Ecallantide-Kalbitor®
Ecallantide (DX-88) is a recombinant protein of 60 amino acids that reversibly inhibits kallikrein activity [26]. The recommended dose is 10 mg up to 3 times a day subcutaneously with a maximum of two doses in 24 hours. Currently it is not approved for use in children, but there are published studies in which part of the recruited patients are children, such as EDEMA 3 study in which are involving 71 patients with a minimum age of 10 years [27]. The most common side effects are headache, nausea, fever, diarrhea, nasopharyngitis or local reactions at the injection site [26-29] (Figure 4).
Androgens
In many countries the use of derivatives of androgens (methyltestosterone, danazol, stanozolol and oxandrolone) are used in therapies short and long term prophylaxis. The recommended dose for short treatments are 2.5 to 10 mg/kg daily (maximum 600 mg/day) for 5 days, two days before the intervention [15]. Currently its use is not recommended in children as long-term prophylaxis [15,30,31]. No clinical studies in children and adolescents with HAE. A few cases cohort provides data on the efficacy and tolerability at low doses [32].
Antifibrinolytics
Antifibrinolytic (ε-aminocaproic acid and tranexamic acid) are chemically synthesized and perform their action in inhibiting HAE conversion of plasminogen to plasmin. They are less effective than androgens [15]. The recommended dose tranexamic acid is 20-50 mg/kg/day. Protocol acute attacks of HAE deficit of C1-INH (type 1 and 2) (Figure 5).
Short-term Prophylaxis
It target is to prevent potential AE episodes triggered by diagnostic or therapeutic processes. It is indicated in all procedures involving trauma in the cervicofacial region, risk of edema of upper airway, and any diagnostic or therapeutic to prevent local edema in the work area not to alter the outcome of the proceeding procedure. Any patient receiving short-term prophylaxis should be monitored and be available for treating acute episode for 48 hours after the procedure, since the risk of angioedema not totally canceled (Figure 6).
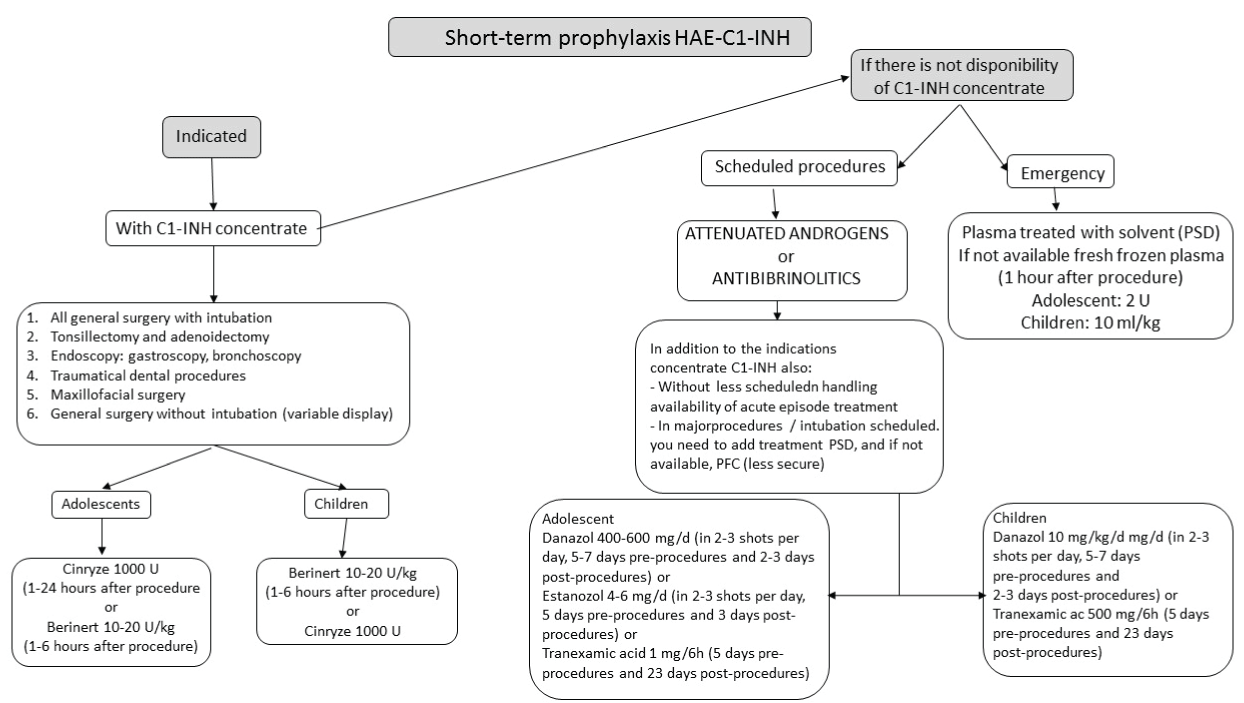
Currently, except that the procedure is less handling, with availability of specific treatment for acute episode, C1-INH concentrates are the treatment of choice. In case of unavailability of C1-INH, other alternatives are attenuated androgens (danazol, stanozolol) or antifibrinolytic (tranexamic acid, epsilon amino caproic acid) in elective procedures, or plasma treated with solvent, if not available fresh frozen plasma, in case of emergency.
Long-term Prophylaxis
It should be considered individually by frequency, anatomical location and other special situations. It is indicated in patients with more than one attack per month or whose duration exceeds 24 days a year with AE. It should be considered in patients who have recently undergone upper airway edema or a serious attack on abdominal or head and neck level. The remaining situations where long-term prophylaxis can make are: attacks of rapid progression, poor control with demand treatment for acute attacks and overuse of medical resources (> 3 visits to emergency services, > 1 hospitalization, ICU admission in one year), unavailability of acute treatment or difficult access to primary care, high burden of disease, impaired quality of life and serious injuries spontaneous (Figure 7).
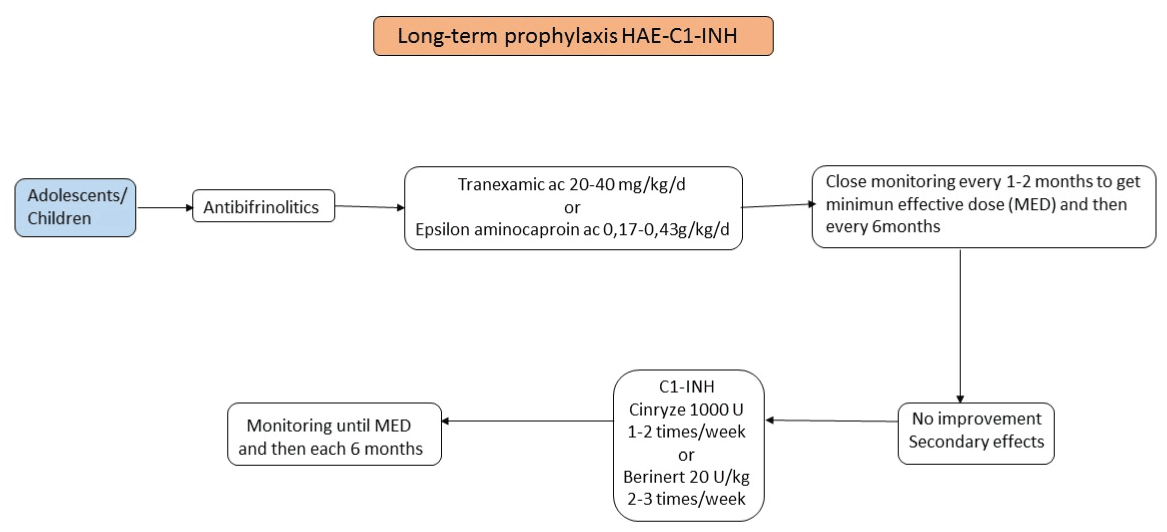
Unlike short-term prophylaxis, the long-term prophylaxis should be flexible taking into account the availability of specific drugs, distance to hospital, etc. Currently, the first-line treatment involves the administration of antifibrinolytic in children under 18 and adults attenuated androgens.
If no improvement or side effects, switching between them or the administration of C1-INH concentrate is valued.
Discussion
The hereditary angioedema disease remains unknown to most doctors who treat patients in emergency, especially for pediatricians because its incidence is low among most of the pathologies treated daily in the units of primary and hospital care; indeed, even in the presence of patients with clinical suspicion are other diagnostic options handle, with the AE one of the most unknown diseases but at increased risk of endangering the lives of patients, or sometimes incapacitating life usually the patient and family.
In the Allergy Pediatric Unit of the University Hospital Puerto Real we have clinical data from several patients, namely 5 children aged between 19 months and 12 years who have suffered at some moment an episode of angioedema. These ages are similar to those presented by most authors, as described in US series for which the average age would be 6 years old [33]. Other authors as Farkas describes series of patients in Hungary and reported an average age of first attacks of 6.6 years, range 4-11 years [34]. Similarly, series in Germany show an average first attack at age 11 years old [35], Denmark 9.5 years [36] and 12.5 years in Spain [37].
The first form of presentation for all patients was similar, starting with erythema affects half of the face, usually with craniocaudally distribution and without overcoming the midline of the face, sometimes extending to the base of the neck in the same side of the face. Our patients do not follow the usual presentation form, because although the symptoms may begin in childhood time, episodes of attack of the disease are rare before puberty, in fact the presence of severe symptoms at an early age will indicate an adverse prognostic factor of prediction in adulthood [32,35,36]. In our case series, we have data from the first attack on an infant 19 months that began with lip swelling, initially associated with insect bites, but after a few hours spread to face the same side without exceeding the midline, and this presentation has been standard in all other cases except one 8 year old male patient who presented with facial involvement and respiratory symptoms of breathlessness and a female patient aged 12, sister of the former, which debuted with cramping abdominal pain, nausea and vomiting, initially diagnosed as acute gastroenteritis (until then the C1-INH deficiency was unknown), and evolved with decay, dehydration, tachycardia to detect the presence of C1-INH deficiency. In the differential diagnosis the possibility of appendicitis, mesenteric lymphadenitis, intussusception, intestinal torsion intestinal strangulation and others who come to also assess perforated Meckel's diverticulum, of polycystic ovary syndrome (typical of women in the final stage of the suggested raised puberty or adult), and even bleeding, heart attack or ovarian torsion [38-40], easily identified by ultrasonography techniques [41,42].
In all those cases, the attacks are not accompanied by fever and laboratory markers of inflammation are normal. We can detect increased levels of prothrombin (fragments 1 + 2) and D dimers which we can use as objective markers, but not as specific biomarkers [43].
Although clinical data are suggestive of acute angioedema crisis, these symptoms can occur in other locations including urinary system, urethra, muscles and joints, liver, pericardium, pleura or even neurological symptoms [4,35].
The duration of the crisis in our patients have an average of seven days, in contrast to most authors. In our series, this time presents an increased variation due to the two previously mentioned brothers because they pointed jugular intravenous drug treatment for the crisis. In the case of the female patient, the crisis gave way after a dose of Berinert IV 800 U (20 U/kg) without additional dose, just prior to the unavailability of data from HAE, treatment management it took four days. But the brother of eight years, to debut with respiratory disease two weeks after the attack of his sister and knowing the background, so the diagnosis was immediately, we started using a first dose of 600 U iv Berinert ( 20 U/kg), after reassessing the patient's progress and persist shortness of breath and impaired airway accompanied by symptoms of discomfort when swallowing oral liquid after six hours, we proceeded to administer a second dose of Berinert IV 600 U that was not effective, so we finally decided to associate the third dose of medicine Cinryze 1000 U iv slowly, eventually checking the resolution of the HAE attack after 3 hours of the first dose of Cinryze.
After evaluating the current literature we found no cases of resistance to Berinert in children so that we can establish that it is the first time that Cinryze is administered, not as a first line treatment but as rescue therapy for failure Berinert, although there is a study of use of Cinryze in children, as the study published by Lumry in which comparing three doses of Cinryze in children 2 to 12 years and weighing more than 10 kg, 500 U (if the patient weighed 10-25 k ), 1000 U and 1500 U (if the weight was > 25 kg) with well tolerating in all three cases [44,45]. The cause has not been found, since both drugs work in the same location in the path of classical complement activation although we suspect that nanofiltration which is subjected human plasma for the Cinryze allows its functionality more pure and block more activation quickly C3 to C3a, but another reason may be that the male patient in eight years the C1-INH levels were almost undetectable in laboratory tests, specifically 0.9 mg/dl while in the other patients ranged between 5.77 and 13.5 mg/dl.
Both patients are currently stable without crisis HAE attack for more than 6 months, in the case of the female patient has Berinert at home while brother has Cinryze so if you have an acute attack, medical services that come to his home to begin the administration of both drugs before and during transport to hospital. We proceeded to carry out genetic studies in some patients obtained so far only patient outcome younger age, the infant of 19 months, making the C164 variant of T (Phe33fsX56) in the gene SERPING1.
WAO Generals' Recommendations
Finally, this article cannot finish to say nothing of the recommendations prescribed by WAO for the general indications, adults and child with HAE [46].
References
-
Agostoni A, Aygören-Pürsün E, Binkley KE, Blanch A, Bark K, et al. (2004) Hereditary and acquired angioedema: problems and progress: proceedings of the third C1 esterase inhibitor deficiency workshop and beyond. J Allergy ClinImmunol 114: S51-131.
-
Zuraw BL (2008) Clinical practice. Hereditary angioedema. N Engl J Med 359: 1027-1036.
-
Velasco-Medina AA, Cortés-Morales G, Barreto-Sosa A, Velázquez-Sámano G (2011) Pathophysiology and advances in the treatment of Hereditary Angioedema. Rev Alerg Mex 58: 112-119.
-
Bas M, Adams V, Suvorava T, Niehues T, Hoffmann TK, et al. (2007) Nonallergic angioedema: role of bradykinin. Allergy 62: 842-856.
-
Davis AE (2008) Hereditary angioedema: a current state-of-the-art review, III: mechanisms of hereditary angioedema. Ann Allergy Asthma Immunol 100: S7-12.
-
Kaplan AP, Joseph K (2010) The bradykinin-forming cascade and its role in hereditary angioedema. Ann Allergy Asthma Immunol 104: 193-204.
-
Bruce L, Zuraw MD (2008) Hereditary Angioedema. N Eng l J Med 359:1027-1036
-
Prematta MJ, Bewtra AK, Levy RJ, Wasserman RL, Jacobson KW, et al. (2012) Per-attack reporting of prodromal symptoms concurrent with C1-inhibitor treatment of hereditary angioedema attacks. Adv Ther 29: 913-922.
-
Temiño VM, Peebles RS Jr (2008) The spectrum and treatment of angioedema. Am J Med 121: 282-286.
-
Cicardi M, Zanichelli A (2010) Angioedema due to C1 inhibitor deficiency in 2010. Intern Emerg Med 5: 481-486.
-
Gompels MM, Lock RJ, Abinun M, Bethune CA, Davies G, et al. (2005) C1 inhibitor deficiency: consensus document. Clin Exp Immunol 139: 379-394.
-
Caballero T (2011) Consensus Statement on the diagnosis, management and treatment of angioedema mediated by bradykinin. Part I. Classification, epidemiology, pathophysiology, genetics, clinical symptoms and diagnosis. J InvestigClinImmunol 21: 333-347.
-
Pasto Cardona L, Bordas Orpinell J, Mercadal Orfila G, Pérez de la Vara A, Jodar Massanés R (2003) Profhylaxis and treatment of hereditary and acquired angioedema at HUB; use of the C1-esterase inhibitor. Farm Hosp 27: 346-352.
-
Vitrat-Hincky V, Gompel A, Dumestre-Perard C, Boccon-Gibod I, Drouet C, et al. (2010) Type III hereditary angio-oedema: clinical and biological features in a French cohort. Allergy 65: 1331-1336.
-
Bowen T, Cicardi M, Farkas H (2010) International Consensus algorithm for the diagnosis, therapy and management of hereditary angioedema. Allergy Asthma ClinImmunol 6-24.
-
Boyle RJ, Nikpour M, Tang ML (2005) Hereditary angio-oedema in children: a management guideline. Pediatr Allergy Immunol 16: 288-294.
-
Craig TJ, Levy RJ, Wasserman RL, Bewtra AK, Hurewitz D, et al. (2009) Efficacy of human C1 esterase inhibitor concentrate compared with placebo in acute hereditary angioedema attacks. J Allergy Clin Immunol 124: 801-808.
-
Craig TJ, Bewtra AK, Bahna SL, Hurewitz D, Schneider LC, et al. (2011) C1 esterase inhibitor concentrate in 1085 Hereditary Angioedema attacks--final results of the I.M.P.A.C.T.2 study. Allergy 66: 1604-1611.
-
Kreuz W, Rusicke E, Martinez-Saguer I, Aygören-Pürsün E, Heller C, et al. (2012) Home therapy with intravenous human C1-inhibitor in children and adolescents with hereditary angioedema. Transfusion 52: 100-107.
-
Martinez-Saguer I, Rusicke E, Aygören-PürsünE, von Hemtig N, Klingebiel T, et al. (2010) Pharmacokinetic analysis of human plasma-derived pasteurized C1-inhibitor concentrate in adults and children with hereditary angioedema; a prospective study. Transfusion 50: 354-360.
-
Zuraw BL, Busse PJ, White M, Jacobs J, Lumry W, et al. (2010) Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema. N Engl J Med 363: 513-522.
-
Tallroth GA (2011) Long-term prophylaxis of hereditary angioedema with a pasteurized C1 inhibitor concentrate. Int Arch Allergy Immunol 154: 356-359.
-
Vinhas de Souza M, Keller-Stanislawski B, Blake K, Hidalgo-Simon A, Arlett P, et al. (2012) Drug-induced PML: a global agenda for a global challenge. Clin Pharmacol Ther 91: 747-750.
-
Frank MM, Jiang H (2008) New therapies for hereditary angioedema: disease outlook changes dramatically. J Allergy Clin Immunol 121: 272-280.
-
Zuraw B, Cicardi M, Levy RJ, Nuijens JH, Relan A, et al. (2010) Recombinant human C1-inhibitor for the treatment of acute angioedema attacks in patients with hereditary angioedema. J Allergy Clin Immunol 126: 821-827.
-
Garnock-Jones KP (2010) Ecallantide: in acute hereditary angioedema. Drugs 70: 1423-1431.
-
Cicardi M, Levy RJ, McNeil DL, Li HH, Sheffer AL, et al. (2010) Ecallantide for the treatment of acute attacks in hereditary angioedema. N Engl J Med 363: 523-531.
-
Levy RJ, Lumry WR, McNeil DL, Li HH, Campion M, et al. (2010) EDEMA4: a phase , double-blind study of subcutaneous ecallantide treatment for acute attacks of hereditary angioedema. Ann Allergy Asthma Immunol 104: 523-529.
-
Sheffer AL, Campion M, Levy RJ, Li HH, Horn PT, et al. (2011) Ecallantide (DX-88) for acute hereditary angioedema attacks: integrated analysis of 2 double-blind, phase 3 studies. J Allergy Clin Immunol 128: 153-159.
-
Cicardi M, Bork K, Caballero T (2012) Evidence-based recommendations for therapeutic management of angioedema owing to hereditary C1 inhibitor deficiency: consensus report of an International Working Group. Allergy 67: 147-157.
-
Maurer M, Magerl M (2010) Hereditary angioedema: an update on available therapeutic options. J Dtsch Dermatol Ges 8: 663-672.
-
Farkas H, Harmat G, Gyeney L, Füst G, Varga L (1999) Danazol therapy for hereditary angio-oedema in children. Lancet 354: 1031-1032.
-
Donaldson VH, Rosen FS (1966) Hereditary angioneurotic edema: a clinical survey. Pediatrics 37: 1017-1027.
-
Farkas H (2010) Pediatric hereditary angioedema due to C1-inhibitor deficiency. Allergy Asthma Clin Immunol 6: 18.
-
Bork K, Meng G, Staubach P, Hardt J (2006) Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med 119: 267-274.
-
Bygum A, Fagerberg CR, Ponard D, Monnier N, Lunardi J, et al. (2011) Mutational spectrum and phenotypes in Danish families with hereditary angioedema because of C1 inhibitor deficiency. Allergy 66: 76-84.
-
Roche O, Blanch A, Caballero T, Sastre N, Callejo D, et al. (2005) Hereditary angioedema due to C1 inhibitor deficiency: patient registry and approach to the prevalence in Spain. Ann Allergy Asthma Immunol 94: 498-503.
-
Lindecken KD, Adolph M, Paterakis S (1991) Pedicle torsion, hemorrhagic ovarian infarct. A rare cause of pediatric acute abdomen. Zentralbl Chir 116: 679-682.
-
Foix-L´HeliasL, Weiss L, Mollet-Boudjemline A, Fallik D, Trioche-Eberschweiler P, et al. (2005) Recurring acute abdominal pains in an adolescent as the presenting manifestations of hereditary angioneuroticedema. ActaPaediatr 945: 1158-1161.
-
Pritzker HA, Levin TL, Weinberg G (2004) Recurrent colocolic intussusception in a child with hereditary angioneurotic edema: reduction by air enema. J Pediatr Surg 39: 1144-1146.
-
Farkas H, Harmat G, Fekete B, Karádi I, Visy B, et al. (2002) Acute abdominal attack of hereditary angioneurotic oedema associated with ultrasound abnormalities suggestive of acute hepatitis. Acta Paediatr 91: 971-974.
-
Farkas H, Harmat G, Kaposi PN, Karádi I, Fekete B, et al. (2001) Ultrasonography in the diagnosis and monitoring of ascites in acute abdominal attacks of hereditary angioneurotic oedema. Eur J Gastroenterol Hepatol 13: 1225-1230.
-
Cugno M, Zanichelli A, Bellatorre AG, Griffini S, Cicardi M (2009) Plasma biomarkers of acute attacks in patients with angioedema due to C1-inhibitor deficiency. Allergy 64: 254-257.
-
Lumry W, Soteres D, Gower R, Jacobson KW, Li HH, et al. (2015) Safety and efficacy of C1 esterase inhibitor for acute attacks in children with hereditary angioedema. Pediatr Allergy Immunol 26: 674-680.
-
Baeza ML, Caballero Molina T, Crespo Diz C, González-Quevedo, Guilarte Clavero M, et al. (2013) Algorithm for diagnosis and treatment of hereditary angioedema as a tool for management. Farm Hosp 37: 521-529.
-
Craig T, Aygören-Pürsün E, Bork K, Bowen T, Boysen H, et al. (2012) WAO Guideline for the Management of Hereditary Angioedema. World Allergy Organ J 5: 182-199.
